Two French genotypes of soybean (Glycine max L.) belonging to the same maturity group I were selected for their similar root or aerial biomasses and their contrasted root system architecture: Stocata (obtained from RAGT Semences) and Wendy (obtained from Caussade Semences) (Maslard et al. 2021). Seeds were calibrated and pre-germinated for 4 days at 20 °C in a Fitoclima S600 germinator (Aralab, Rio de Mouro, Portugal) before transplantation. Seedlings were grown in RhizoTubes® (Figure 7.1) filled with sandy soil, allowing the visualization of the root system (Chapter 7). The soil was collected from a grassland at Flammerans near Dijon (Burgundy). Plants were grown under controlled conditions, in the Plant Phenotyping Platform for Plant and Microorganism Interaction (4PMI) at INRAE in Dijon (France) (47°32’N, 5°02’E) during 23 days in RhizoTube® (Figure 2.1). Two seedlings were transplanted in each of the 144 RhizoTubes®, in two different identical greenhouses (Figure 2.4). Two plants grew in each RhizoTubes® and were destined for four different types of analysis (Figure 2.2). At the same time, each seedling was inoculated with 1mL of Bradyrhizobium diazoefficiens corresponding to 108 rhizobia.
Figure 2.1: Experimental design of the experiment
Figure 2.2: Harvest diagram for the experiment. RT for Rhizotube. Harvest 1 took place 5 days after sowing. Harvest 2 was carried out on the day after sowing. For harvest 1, there are two plants of the same genotype per rhizotube (RT0). For each plant, ecophysiological measurements and the content of elements in each compartment were conducted. For harvest 2, in each rhizotube (RT1, RT2), there were two plants of the same genotype that have undergone the same stress conditions. Ecophysiological measurements and the element content were performed on one of the two plants. The other plant was designated for molecular analyses. Transcriptomic measurements were conducted on a pool of two roots from two plants from two different RhizoTubes (RT1 and RT2) of the same genotype having received the same stress conditions. Metabolomic analyses of leaves, stems, and roots were performed on the same plant combination. The soil near this plant was collected for metabolomic analyses, pH analyses, and microbial community analyses (Rhizosphere) (no assembly of these soils). The soil between these two plants was used for metabolomic analyses as well as for microbial community analyses (Bulk soil). During this experiment, other RhizoTubes (RT3) also containing two plants of the same genotype were in the same greenhouse. One was designated for microbial community analyses, the other for metabolomic measurements of exudates. On the plant used for the root exudates, ecophysiological measurements were also conducted. Root system architecture (RSA) and total evapotranspiration were measured on all RhizoTubes during the experiment. WW: Well Watered; WS: Water Stress; OT: Optimal temperature; HS: Heat Stress.
Air relative humidity was controlled to 45% and the photoperiod was set to 16h thanks to an artificial lighting (PAR of 272µmol.m-2. s-1) supplied with sodium lamps (400W lamp, HPS Plantastar, OSRAM, Munich, Germany). The PAR data as well as the other climatic data (humidity, temperature) are presented below (Figure 2.3).
Every Rhizotube® was autonomously weighed both before and after each watering session, if applicable, three times a day. This process was conducted to establish the evolving water evapotranspiration (ml)(Figure 3.2) and precise solution quantities (Chapter 3). The cumulative evapotranspiration during experiment was computed by summing and halving (accounting for the two plants within each Rhizotube®) the individual daily differences in Rhizotube® weight between consecutive watering events, or weight variation (Chapter 3). A N-free nutrient solution as described in (Voisin et al. 2003) was applied 3 times a day to allow 20% SWC (80% of field capacity) of water content in the RhizoTube® (Figure 3.1). Mean day/night temperatures were 32°C/24°C in two greenhouses (Figure 2.3).
Following the first week of growth, 12 plants per genotype were harvested (Figure 2.2). Subsequently, we implemented a factorial design consisting of eight different conditions (Sto_WW_OT, Sto_WS_OT, Sto_WW_HS, Sto_WS_HS, Wen_WW_OT, Wen_WS_OT, Wen_WW_HS, Wen_WS_HS) involving the two genotypes, Stocata (Sto) and Wendy (Wen), and four climatic conditions, composed of two stress factors. One factor was related to watering, with half of plants being subjected to either well-watered (WW) conditions with a SWC of 20 percent (field capacity of 80%) soil water content of 20%, or water stress (WS) conditions achieved by withholding water to reach 10% soil water content (Figure 3.1). The second factor was related to temperature. One greenhouse was maintained at an optimal temperature (OT) of 32°C/24°C (day/night), while the other greenhouse experienced two progressive heat waves of 40°C for 3 days (Figure 2.3 A), representing heat stress conditions (HS). Five days after sowing some plants were harvested for physiological analysis (including ionomic analysis) (H1). Then 20 days after sowing, (after the period of stress) plant were harvest (H2) and each organ was collected separately (leaves, stems and roots).
Four different types of analysis were performed:
Eight plants per condition were dedicated to ecophysiological analyses (Chapter 3) including ionomics (Chapter 10) (8 plants per condition (only 6 plants over the 8 plants were used for ionomic analysis).
Eight plants per condition were dedicated to transcriptomic and metabolomic analyses. After being harvested in liquid nitrogen, four replicates of two plants each were pooled together for each of the four treatments, for each genotype and each plant compartment (leaf, stem, and root). These pooled samples were then ground for analysis. Only root samples were analysed for transcriptomic analysis (?sec-rnaseq) and all three compartments were analyzed for metabolomics (?sec-metabo). Plant soil located close to or far from the roots was collected for metabolomics (?sec-metabo) analyses as well as for microbial community (?sec-mcom) and pH analyses.
Five plants per condition were dedicated to exudate collection and exudation analyses by metabolomics (?sec-metabo) and to biomass measurement of each compartment.
Five plants per condition were dedicated to the analysis of microbial communities (fungi or bacteria) in different compartments (phylloplane, leaf endosphere, root endosphere, and rhizoplane) (?sec-mcom)
Plants A and B were harvested from the same rhizotube. Plants C and D were harvested from the same rhizotube. All plants were imaged three times a week to measure aerial and root architecture.
2.2 Climate in the greenhouse
Code
#temperature compilationdata_global_climat_select=climat_function()data_global_climat_select_moy=mean_interval_function(data_global_climat_select,5,"min")#graphiqueggplotly(ggplot(data_global_climat_select_moy,aes(x=as.POSIXct(Date_Time),y=Hygrometry,col=CardID))+geom_line()+#geom_point(size=0.5)+facet_grid(~Unit))# Specify cutting interval in minutes (e.g. 60 minutes)interval_minutes <-60data_mean <- data_global_climat_select_moy %>%filter(CardID!="ARIA") %>%mutate(Date_Time =as.POSIXct(Date_Time, format="%Y-%m-%d %H:%M")) %>%filter(Date_Time>as.POSIXct("2021-09-22 10:00:00")& Date_Time<as.POSIXct("2021-10-14 00:00:00")) %>%mutate(interval_id =cut(as.POSIXct(Date_Time),breaks =seq(min(Date_Time), max(Date_Time) + interval_minutes *60, by = interval_minutes *60),labels =FALSE,include.lowest =TRUE )) %>% dplyr::group_by(Unit, interval_id) %>% dplyr::summarise(mean_temp =mean(Temperature, na.rm =TRUE),mean_humidity =mean(Hygrometry, na.rm =TRUE),mean_PAR =mean(PAR, na.rm =TRUE)) %>%ungroup() plot_mean_temperature=data_mean %>%ggplot(aes(x = interval_id/24, y = mean_temp, color = Unit)) +geom_line() +labs(x ="Day", y ="Average temperature (°C)", color ="Unit") +scale_fill_manual(values=temperature_color)+scale_color_manual(values=temperature_color)+ my_theme plot_mean_temperatureplot_mean_humidity=data_mean %>%ggplot(aes(x = interval_id/24, y = mean_humidity, color = Unit)) +geom_line() +labs(x ="Day", y ="Average humidity (%)", color ="Unit") +scale_y_continuous(labels = scales::percent_format(scale =100))+scale_fill_manual(values=temperature_color)+scale_color_manual(values=temperature_color)+ my_themeplot_mean_humidityplot_mean_PAR=data_mean %>%ggplot(aes(x = interval_id/24, y = mean_PAR, color = Unit)) +geom_line() +labs(x ="Day", y=expression(atop("Photosynthetically active",paste( "radiation ", "(µmol/ ",m^-2," /",s^-1,")"))),color ="Unit") +scale_fill_manual(values=temperature_color)+scale_color_manual(values=temperature_color)+ my_themeplot_mean_PARcombined_plot=ggarrange(plot_mean_temperature,plot_mean_humidity,plot_mean_PAR,ncol=1, nrow=3,common.legend =TRUE, legend="right",labels =c("A","B","C"),align='v')# Create a title and subtitlecombined_plot_title=annotate_figure(annotate_figure(combined_plot,top=text_grob("The measurement represents an average each hour derived from four sensors positioned at \n various locations within the unit for each unit.", size =12,hjust=0,x=0.1,y=0.4), ),top=text_grob("Temperature, humidity and PAR during the experiment",face ="bold", size =14,hjust=0,x=0.1,y=0.4)) ; combined_plot_title# exportggsave(here::here("report/intro/plot/combined_plot.svg"), combined_plot_title, width =21, height =25, units ="cm")
Figure 2.3: Temperature (A), humidity (B) and PAR (C) during the experiment. The measurement represents an average each hour derived from four sensors positioned at various locations within the unit for each unit.
Bonnus: Correction for spatial trends over time
2.2.1 Correction for spatial trends over time
If there had been a significant bias, I would have had to take it into account
But according to the model (Chapter 3 and Chapter 6) using biomass there is only a slight effect of position in the greenhouse, and in addition I randomized the experiments.
The first step when modeling platform experiment data with the statgenHTP package is creating an object of class TP (Time Points) For some variable you havent (one harvest). In this object, the time points are split into single data.frames. It is then used throughout the statgenHTP package as input for analyses.
Code
# Formatting the data = create a list with one data frame per time pointU2_all_time_point <-createTimePoints(dat = df_physio %>%filter(unit=="U2") %>%#filter(paire==F) %>% mutate(plant_num=as.character(plant_num)) %>%mutate(position_b =ifelse(as.numeric(plant_num) %%2==1, as.numeric(position) *2-1, as.numeric(position) *2)) %>%mutate(pos=paste0("c",storage_line,"r",position_b)) %>%mutate(timepoints="2021-10-11") %>%mutate(harvest_condition=paste0(recolte, "_", condition)) %>% dplyr::select(timepoints, harvest_condition, position_b, storage_line, sum_biomass,pos,rep),experimentName ="U2_Soybean_2021",genotype ="harvest_condition",timePoint ="timepoints",repId ="rep", plotId ="pos",rowNum ="position_b", colNum ="storage_line",addCheck =FALSE, #if you have some geneotype repeate (that help the model to control)checkGenotypes =c("Wen_WW_OT", "Wen_WW_HS", "Sto_WW_OT", "Sto_WW_HS") )U3_all_time_point <-createTimePoints(dat = df_physio %>%filter(unit=="U3") %>%#filter(paire==F) %>% mutate(plant_num=as.character(plant_num)) %>%mutate(position_b =ifelse(as.numeric(plant_num) %%2==1, as.numeric(position) *2-1, as.numeric(position) *2)) %>%mutate(pos=paste0("c",storage_line,"r",position_b)) %>%mutate(timepoints="2021-10-11") %>%mutate(harvest_condition=paste0(recolte, "_", condition)) %>% dplyr::select(timepoints, harvest_condition, position_b, storage_line, sum_biomass,pos,rep),experimentName ="U3_Soybean_2021",genotype ="harvest_condition",timePoint ="timepoints",repId ="rep", plotId ="pos",rowNum ="position_b", colNum ="storage_line",addCheck =FALSE, #if you have some geneotype repeate (that help the model to control)checkGenotypes =c("Wen_WW_OT", "Wen_WW_HS", "Sto_WW_OT", "Sto_WW_HS") )U2_H2_time_point <-createTimePoints(dat = df_physio %>%filter(unit=="U2") %>%filter(paire==F) %>%filter(recolte==2) %>%mutate(plant_num=as.character(plant_num)) %>%mutate(position_b =ifelse(as.numeric(plant_num) %%2==1, as.numeric(position) *2-1, as.numeric(position) *2)) %>%mutate(pos=paste0("c",storage_line,"r",position_b)) %>%mutate(timepoints="2021-10-11") %>%mutate(harvest_condition=paste0(recolte, "_", condition)) %>% dplyr::select(timepoints, harvest_condition, position, storage_line, sum_biomass,pos,rep),experimentName ="U2_Soybean_2021",genotype ="harvest_condition",timePoint ="timepoints",repId ="rep", plotId ="pos",rowNum ="position", colNum ="storage_line",addCheck =FALSE, #if you have some geneotype repeate (that help the model to control)checkGenotypes =c("Wen_WW_OT", "Wen_WW_HS", "Sto_WW_OT", "Sto_WW_HS") )U3_H2_time_point <-createTimePoints(dat = df_physio %>%filter(unit=="U3") %>%filter(paire==F) %>%filter(recolte==2) %>%mutate(plant_num=as.character(plant_num)) %>%mutate(position_b =ifelse(as.numeric(plant_num) %%2==1, as.numeric(position) *2-1, as.numeric(position) *2)) %>%mutate(pos=paste0("c",storage_line,"r",position)) %>%mutate(timepoints="2021-10-11") %>%mutate(harvest_condition=paste0(recolte, "_", condition)) %>% dplyr::select(timepoints, harvest_condition, position, storage_line, sum_biomass,pos,rep),experimentName ="U3_Soybean_2021",genotype ="harvest_condition",timePoint ="timepoints",repId ="rep", plotId ="pos",rowNum ="position", colNum ="storage_line",addCheck =FALSE, #if you have some geneotype repeate (that help the model to control)checkGenotypes =c("Wen_WW_OT", "Wen_WW_HS", "Sto_WW_OT", "Sto_WW_HS") )
Plot the layout, NA indicate a probleme with the plant. (It has not been harvested)
Code
## Plot the layout for the 3rd time points.p_u2=plot(U2_all_time_point,plotType ="layout",timePoints =c(1),highlight =c("1_Wen_WW_OT", "1_Sto_WW_OT","2_Wen_WW_OT", "2_Wen_WS_OT", "2_Wen_WW_HS", "2_Wen_WS_HS","2_Sto_WW_OT", "2_Sto_WS_OT", "2_Sto_WW_HS", "2_Sto_WS_HS"),showGeno =TRUE)p_u3=plot(U3_all_time_point,plotType ="layout",timePoints =c(1),highlight =c("1_Wen_WW_OT", "1_Sto_WW_OT","2_Wen_WW_OT", "2_Wen_WS_OT", "2_Wen_WW_HS", "2_Wen_WS_HS","2_Sto_WW_OT", "2_Sto_WS_OT", "2_Sto_WW_HS", "2_Sto_WS_HS"),showGeno =TRUE)svg(file=here::here("report/intro/plot/layout_unit_U2.svg"),width =9, height =9)p_u2dev.off()svg(file=here::here("report/intro/plot/layout_unit_U3.svg"),width =9, height =9)p_u3dev.off()# # Check the heatmap of the raw data at one time point# plot(U2_time_point,# plotType = "layout",# timePoints = 1,# traits = "sum_biomass")
(a) U2
(b) U3
Figure 2.4: Layout of Rhizotube and plants in the two greenhouses
Code
Trait <-"sum_biomass"spatial_correction<-function(Trait,data_x){ pheno <- data_x[[1]] %>%drop_na(Trait) #%>% filter(genotype %in% c( "2_Wen_WS_OT","2_Sto_WS_OT")) %>% mutate(genotype==substr(genotype, 3, 5)) # select the nb of segments: here all the rows and columns nseg =c(nlevels(pheno$colId), nlevels(pheno$rowId))cat("Time Point: ", timePoint, "\n", file = output_file) # add timePoint numberprint(timePoint) fit.SpATS <-SpATS(response = Trait, # fit the SpATS modelfixed =~ repId,random =~ colId + rowId,spatial =~PSANOVA(colNum, rowNum,nseg = nseg,nest.div =c(2, 2)),genotype ="genotype",genotype.as.random =TRUE,data = pheno,control =list(maxit =50,tolerance =1e-03,monitoring =0)) summary_output <-capture.output(summary(fit.SpATS)) # Captures model summary output all_summaries <-c(all_summaries, paste0("Time Point: ", timePoint, "\n", summary_output))# Add output to variablewriteLines(all_summaries, con = output_file)cat("Traitement pour le trait", Trait, "terminé.\n")plot(fit.SpATS, spaTrend ="percentage")## plot of the spats model with spatial trend as percentage modPhenoSpCheck <-fitModels(TP = data_x, trait = Trait,useRepId =TRUE,useCheck =FALSE,timePoints =1) px<-plot(modPhenoSpCheck, timePoints =1,plotType ="spatial",spaTrend ="percentage")## Extract the corrected values for one time point spatCorrSp <-getCorrected(modPhenoSpCheck, timePoints = timePoint) varianceSp <-getVar(modPhenoSpCheck) ## Extract model components: heritSp <-getHerit(modPhenoSpCheck) effDimSp <-getEffDims(modPhenoSpCheck)return(list(list(plot = px, datacorrect = spatCorrSp)))}result_U2=spatial_correction(Trait ="sum_biomass",data_x = U2_H2_time_point)result_U3=spatial_correction(Trait ="sum_biomass",data_x = U3_H2_time_point)p1<-result_U2[[1]]$plot$`2021-10-11`$p4p2<-result_U3[[1]]$plot$`2021-10-11`$p4p1=p1+labs(title =paste0("Filtted Spatial Trend Unit 2", " ", "sum_biomass"))p2=p2+labs(title =paste0("Filtted Spatial Trend Unit 3" , " ", "sum_biomass"))ggsave(here::here("report/intro/plot/ggcoef_model.svg"),p1+p2)result_all=rbind(result_U2[[1]]$"datacorrect",result_U3[[1]]$"datacorrect")p1=result_all %>%ggplot(aes(x=genotype,y=sum_biomass, fill=genotype))+geom_boxplot()+labs(title="Without correction for sum biomass")p2=result_all %>%ggplot(aes(x=genotype,y=sum_biomass_corr, fill=genotype))+geom_boxplot()+labs(title="With correction for sum biomass")ggsave(here::here("report/intro/plot/comparison.svg"),p1+p2)
Maslard, Corentin, Mustapha Arkoun, Christophe Salon, and Marion Prudent. 2021. “Root Architecture Characterization in Relation to Biomass Allocation and Biological Nitrogen Fixation in a Collection of European Soybean Genotypes.”OCL 28: 48. https://doi.org/10.1051/ocl/2021033.
Voisin, A. S., Salon, Christophe, Jeudy, Christian, and F. R. Warembourg. 2003. “Symbiotic N2 Fixation Activity in Relation to c Economy of Pisum Sativum l. As a Function of Plant Phenology.”Journal of Experimental Botany, 6.992, 54 (393): 2733–44. https://doi.org/10.1093/jxb/erg290.
Source Code
---output: html_documenteditor_options: chunk_output_type: consoleeditor: markdown: wrap: sentence---## Experimental design and climate condition```{r, warning=FALSE}#pkg library(dplyr)library(ggplot2)library(statgenHTP) #remotes::install_github("Biometris/statgenHTP", ref = "develop", dependencies = TRUE)library(SpATS)library(tidyr)library(patchwork)library(readxl)library(plotly)library(lubridate)library(gridExtra)library(ggpubr)# src source(here::here("src/function/card_compilation.R"))# cosmeticstemperature_color=read_excel(here::here("data/color_palette.xlsm")) %>% filter(set == "greenhouse") %>% dplyr::select(color, treatment) %>% pull(color) %>% setNames(read_excel(here::here("data/color_palette.xlsm")) %>% filter(set == "greenhouse") %>% pull(treatment) )my_theme=theme_classic()+theme( plot.title = element_text(size = 14, face = "bold"), axis.title = element_text(size = 12), axis.text = element_text(size = 10), legend.text = element_text(size = 10), panel.grid.major = element_blank(), panel.grid.minor = element_blank())```## Experimental designTwo French genotypes of soybean (Glycine max L.) belonging to the same maturity group I were selected for their similar root or aerial biomasses and their contrasted root system architecture: Stocata (obtained from RAGT Semences) and Wendy (obtained from Caussade Semences) [@maslard2021].Seeds were calibrated and pre-germinated for 4 days at 20 °C in a Fitoclima S600 germinator (Aralab, Rio de Mouro, Portugal) before transplantation.Seedlings were grown in RhizoTubes® (@fig-rhizotube) filled with sandy soil, allowing the visualization of the root system (@sec-root_archicture).The soil was collected from a grassland at Flammerans near Dijon (Burgundy).Plants were grown under controlled conditions, in the Plant Phenotyping Platform for Plant and Microorganism Interaction (4PMI) at INRAE in Dijon (France) (47°32'N, 5°02'E) during 23 days in RhizoTube® (@fig-experimantal_design).Two seedlings were transplanted in each of the 144 RhizoTubes®, in two different identical greenhouses (@fig-greenhouse).Two plants grew in each RhizoTubes® and were destined for four different types of analysis (@fig-harvest_design).At the same time, each seedling was inoculated with 1mL of *Bradyrhizobium diazoefficiens* corresponding to 108 rhizobia.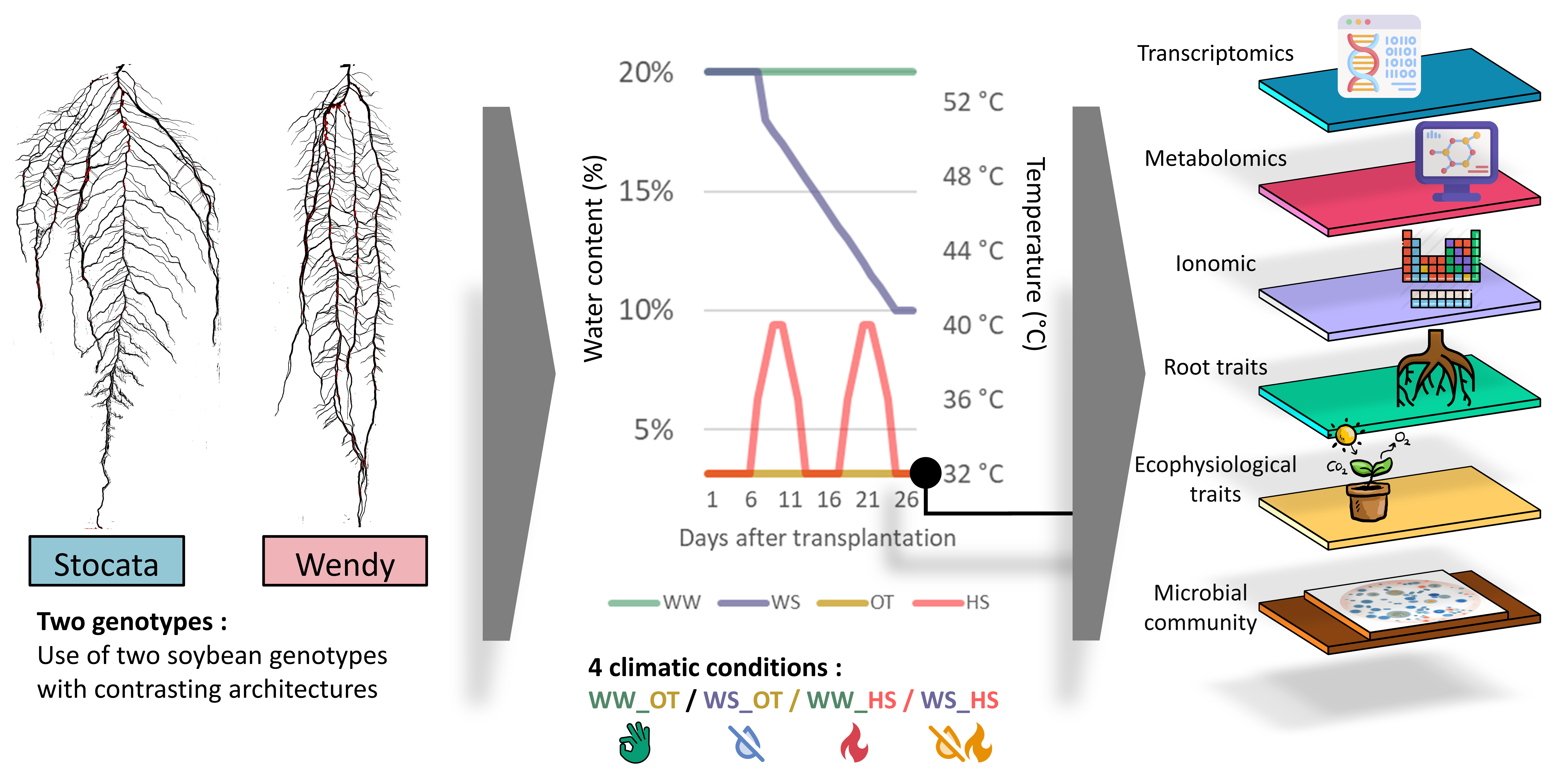{#fig-experimantal_design}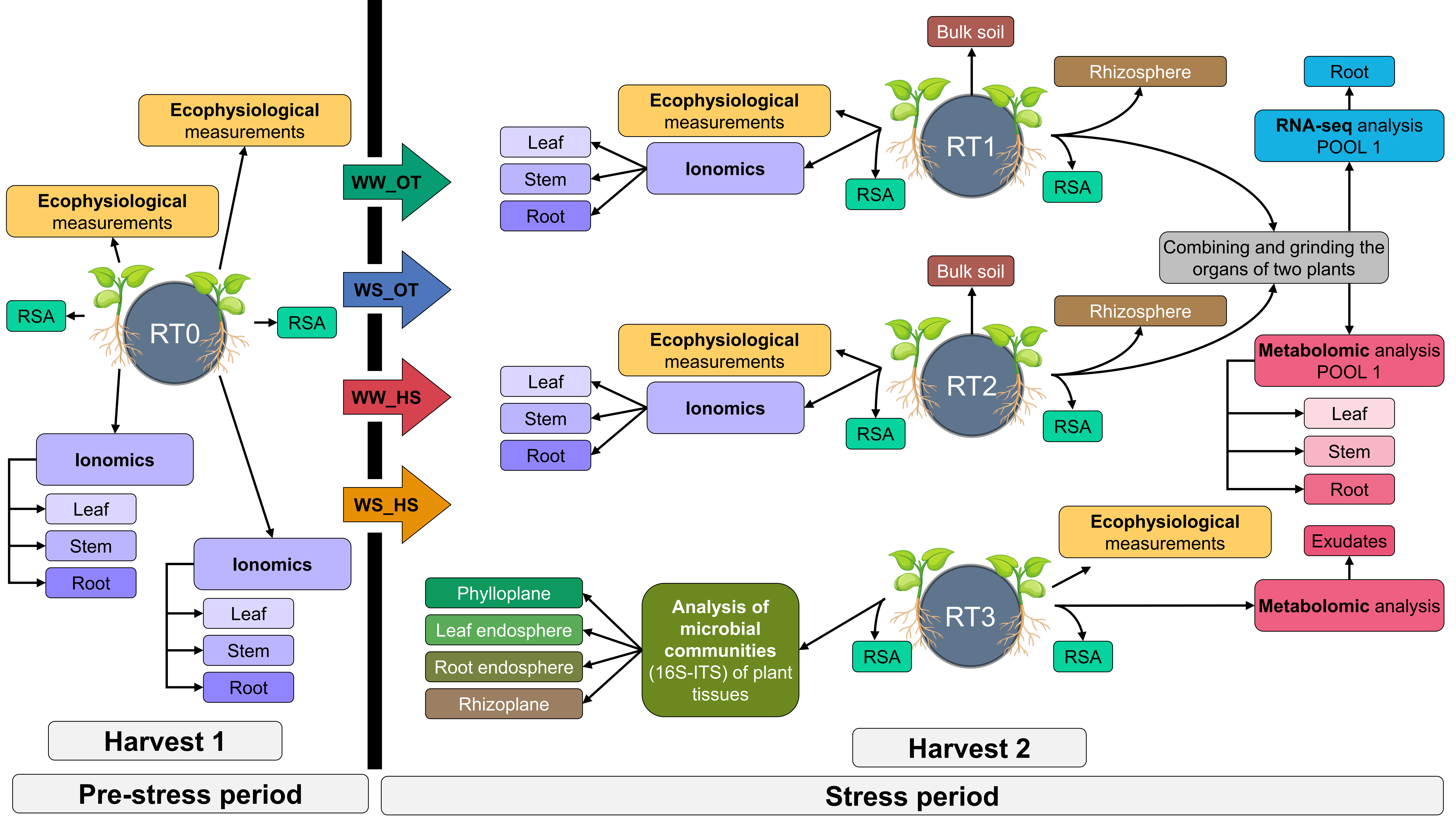{#fig-harvest_design}Air relative humidity was controlled to 45% and the photoperiod was set to 16h thanks to an artificial lighting (PAR of 272µmol.m-2. s-1) supplied with sodium lamps (400W lamp, HPS Plantastar, OSRAM, Munich, Germany).The PAR data as well as the other climatic data (humidity, temperature) are presented below (@fig-metadata).Every Rhizotube® was autonomously weighed both before and after each watering session, if applicable, three times a day.This process was conducted to establish the evolving water evapotranspiration (ml)(@fig-evapotranspiration) and precise solution quantities (@sec-physio).The cumulative evapotranspiration during experiment was computed by summing and halving (accounting for the two plants within each Rhizotube®) the individual daily differences in Rhizotube® weight between consecutive watering events, or weight variation (@sec-physio).A N-free nutrient solution as described in [@voisin_symbiotic_2003] was applied 3 times a day to allow 20% SWC (80% of field capacity) of water content in the RhizoTube® (@fig-water_content).Mean day/night temperatures were 32°C/24°C in two greenhouses (@fig-metadata).Following the first week of growth, 12 plants per genotype were harvested (@fig-harvest_design).Subsequently, we implemented a factorial design consisting of eight different conditions (Sto_WW_OT, Sto_WS_OT, Sto_WW_HS, Sto_WS_HS, Wen_WW_OT, Wen_WS_OT, Wen_WW_HS, Wen_WS_HS) involving the two genotypes, Stocata (Sto) and Wendy (Wen), and four climatic conditions, composed of two stress factors.One factor was related to watering, with half of plants being subjected to either well-watered (WW) conditions with a SWC of 20 percent (field capacity of 80%) soil water content of 20%, or water stress (WS) conditions achieved by withholding water to reach 10% soil water content (@fig-water_content).The second factor was related to temperature.One greenhouse was maintained at an optimal temperature (OT) of 32°C/24°C (day/night), while the other greenhouse experienced two progressive heat waves of 40°C for 3 days (@fig-metadata A), representing heat stress conditions (HS).Five days after sowing some plants were harvested for physiological analysis (including ionomic analysis) (H1).Then 20 days after sowing, (after the period of stress) plant were harvest (H2) and each organ was collected separately (leaves, stems and roots).**Four different types of analysis were performed:**A) Eight plants per condition were dedicated to ecophysiological analyses (@sec-physio) including ionomics (@sec-iono) (8 plants per condition (only 6 plants over the 8 plants were used for ionomic analysis).B) Eight plants per condition were dedicated to transcriptomic and metabolomic analyses. After being harvested in liquid nitrogen, four replicates of two plants each were pooled together for each of the four treatments, for each genotype and each plant compartment (leaf, stem, and root). These pooled samples were then ground for analysis. Only root samples were analysed for transcriptomic analysis (@sec-rnaseq) and all three compartments were analyzed for metabolomics (@sec-metabo). Plant soil located close to or far from the roots was collected for metabolomics (@sec-metabo) analyses as well as for microbial community (@sec-mcom) and pH analyses.C) Five plants per condition were dedicated to exudate collection and exudation analyses by metabolomics (@sec-metabo) and to biomass measurement of each compartment.D) Five plants per condition were dedicated to the analysis of microbial communities (fungi or bacteria) in different compartments (phylloplane, leaf endosphere, root endosphere, and rhizoplane) (@sec-mcom)Plants A and B were harvested from the same rhizotube.Plants C and D were harvested from the same rhizotube.All plants were imaged three times a week to measure aerial and root architecture.## Climate in the greenhouse```{r, eval=F}#temperature compilationdata_global_climat_select=climat_function()data_global_climat_select_moy=mean_interval_function(data_global_climat_select,5,"min")#graphiqueggplotly(ggplot(data_global_climat_select_moy,aes(x=as.POSIXct(Date_Time),y=Hygrometry,col=CardID))+geom_line()+#geom_point(size=0.5)+ facet_grid(~Unit))# Specify cutting interval in minutes (e.g. 60 minutes)interval_minutes <- 60data_mean <- data_global_climat_select_moy %>% filter(CardID!="ARIA") %>% mutate(Date_Time =as.POSIXct(Date_Time, format="%Y-%m-%d %H:%M")) %>% filter(Date_Time>as.POSIXct("2021-09-22 10:00:00")& Date_Time<as.POSIXct("2021-10-14 00:00:00")) %>% mutate(interval_id = cut( as.POSIXct(Date_Time), breaks = seq(min(Date_Time), max(Date_Time) + interval_minutes * 60, by = interval_minutes * 60), labels = FALSE, include.lowest = TRUE )) %>% dplyr::group_by(Unit, interval_id) %>% dplyr::summarise(mean_temp = mean(Temperature, na.rm = TRUE), mean_humidity = mean(Hygrometry, na.rm = TRUE), mean_PAR = mean(PAR, na.rm = TRUE)) %>% ungroup() plot_mean_temperature=data_mean %>% ggplot(aes(x = interval_id/24, y = mean_temp, color = Unit)) + geom_line() + labs(x = "Day", y = "Average temperature (°C)", color = "Unit") + scale_fill_manual(values=temperature_color)+ scale_color_manual(values=temperature_color)+ my_theme plot_mean_temperatureplot_mean_humidity=data_mean %>% ggplot(aes(x = interval_id/24, y = mean_humidity, color = Unit)) + geom_line() + labs(x = "Day", y = "Average humidity (%)", color = "Unit") + scale_y_continuous(labels = scales::percent_format(scale = 100))+ scale_fill_manual(values=temperature_color)+ scale_color_manual(values=temperature_color)+ my_themeplot_mean_humidityplot_mean_PAR=data_mean %>% ggplot(aes(x = interval_id/24, y = mean_PAR, color = Unit)) + geom_line() + labs(x = "Day", y=expression(atop("Photosynthetically active",paste( "radiation ", "(µmol/ ",m^-2," /",s^-1,")"))), color = "Unit") + scale_fill_manual(values=temperature_color)+ scale_color_manual(values=temperature_color)+ my_themeplot_mean_PARcombined_plot=ggarrange(plot_mean_temperature,plot_mean_humidity,plot_mean_PAR,ncol=1, nrow=3,common.legend = TRUE, legend="right",labels = c("A","B","C"),align='v')# Create a title and subtitlecombined_plot_title=annotate_figure( annotate_figure(combined_plot, top=text_grob("The measurement represents an average each hour derived from four sensors positioned at \n various locations within the unit for each unit.", size = 12,hjust=0,x=0.1,y=0.4), ), top=text_grob("Temperature, humidity and PAR during the experiment",face = "bold", size = 14,hjust=0,x=0.1,y=0.4)) ; combined_plot_title# exportggsave(here::here("report/intro/plot/combined_plot.svg"), combined_plot_title, width = 21, height = 25, units = "cm")```{#fig-metadata}<details><summary style="color: #007bff;">Bonnus: Correction for spatial trends over time</summary>### Correction for spatial trends over time::: callout-tip## If there had been a significant bias, I would have had to take it into accountBut according to the model (@sec-physio and @sec-physio_resum) using biomass there is only a slight effect of position in the greenhouse, and in addition I randomized the experiments.:::##### Data importation```{r}df_physio<-read.csv2(here::here("data/physio/global_physio.csv")) %>%mutate(across(c("genotype","water_condition","heat_condition"), as.factor)) %>%mutate(across(c("storage_line","position"), as.integer)) %>%mutate(water_condition=relevel(water_condition, "WW")) %>%mutate(heat_condition=relevel(heat_condition, "OT"))```##### Formatting and visualizing the dataThe first step when modeling platform experiment data with the statgenHTP package is creating an object of class TP (Time Points) *For some variable you havent (one harvest)*.In this object, the time points are split into single data.frames.It is then used throughout the statgenHTP package as input for analyses.```{r}# Formatting the data = create a list with one data frame per time pointU2_all_time_point <-createTimePoints(dat = df_physio %>%filter(unit=="U2") %>%#filter(paire==F) %>% mutate(plant_num=as.character(plant_num)) %>%mutate(position_b =ifelse(as.numeric(plant_num) %%2==1, as.numeric(position) *2-1, as.numeric(position) *2)) %>%mutate(pos=paste0("c",storage_line,"r",position_b)) %>%mutate(timepoints="2021-10-11") %>%mutate(harvest_condition=paste0(recolte, "_", condition)) %>% dplyr::select(timepoints, harvest_condition, position_b, storage_line, sum_biomass,pos,rep),experimentName ="U2_Soybean_2021",genotype ="harvest_condition",timePoint ="timepoints",repId ="rep", plotId ="pos",rowNum ="position_b", colNum ="storage_line",addCheck =FALSE, #if you have some geneotype repeate (that help the model to control)checkGenotypes =c("Wen_WW_OT", "Wen_WW_HS", "Sto_WW_OT", "Sto_WW_HS") )U3_all_time_point <-createTimePoints(dat = df_physio %>%filter(unit=="U3") %>%#filter(paire==F) %>% mutate(plant_num=as.character(plant_num)) %>%mutate(position_b =ifelse(as.numeric(plant_num) %%2==1, as.numeric(position) *2-1, as.numeric(position) *2)) %>%mutate(pos=paste0("c",storage_line,"r",position_b)) %>%mutate(timepoints="2021-10-11") %>%mutate(harvest_condition=paste0(recolte, "_", condition)) %>% dplyr::select(timepoints, harvest_condition, position_b, storage_line, sum_biomass,pos,rep),experimentName ="U3_Soybean_2021",genotype ="harvest_condition",timePoint ="timepoints",repId ="rep", plotId ="pos",rowNum ="position_b", colNum ="storage_line",addCheck =FALSE, #if you have some geneotype repeate (that help the model to control)checkGenotypes =c("Wen_WW_OT", "Wen_WW_HS", "Sto_WW_OT", "Sto_WW_HS") )U2_H2_time_point <-createTimePoints(dat = df_physio %>%filter(unit=="U2") %>%filter(paire==F) %>%filter(recolte==2) %>%mutate(plant_num=as.character(plant_num)) %>%mutate(position_b =ifelse(as.numeric(plant_num) %%2==1, as.numeric(position) *2-1, as.numeric(position) *2)) %>%mutate(pos=paste0("c",storage_line,"r",position_b)) %>%mutate(timepoints="2021-10-11") %>%mutate(harvest_condition=paste0(recolte, "_", condition)) %>% dplyr::select(timepoints, harvest_condition, position, storage_line, sum_biomass,pos,rep),experimentName ="U2_Soybean_2021",genotype ="harvest_condition",timePoint ="timepoints",repId ="rep", plotId ="pos",rowNum ="position", colNum ="storage_line",addCheck =FALSE, #if you have some geneotype repeate (that help the model to control)checkGenotypes =c("Wen_WW_OT", "Wen_WW_HS", "Sto_WW_OT", "Sto_WW_HS") )U3_H2_time_point <-createTimePoints(dat = df_physio %>%filter(unit=="U3") %>%filter(paire==F) %>%filter(recolte==2) %>%mutate(plant_num=as.character(plant_num)) %>%mutate(position_b =ifelse(as.numeric(plant_num) %%2==1, as.numeric(position) *2-1, as.numeric(position) *2)) %>%mutate(pos=paste0("c",storage_line,"r",position)) %>%mutate(timepoints="2021-10-11") %>%mutate(harvest_condition=paste0(recolte, "_", condition)) %>% dplyr::select(timepoints, harvest_condition, position, storage_line, sum_biomass,pos,rep),experimentName ="U3_Soybean_2021",genotype ="harvest_condition",timePoint ="timepoints",repId ="rep", plotId ="pos",rowNum ="position", colNum ="storage_line",addCheck =FALSE, #if you have some geneotype repeate (that help the model to control)checkGenotypes =c("Wen_WW_OT", "Wen_WW_HS", "Sto_WW_OT", "Sto_WW_HS") )```Plot the layout, NA indicate a probleme with the plant.(It has not been harvested)```{r, eval=F}## Plot the layout for the 3rd time points.p_u2=plot(U2_all_time_point, plotType = "layout", timePoints = c(1), highlight = c("1_Wen_WW_OT", "1_Sto_WW_OT","2_Wen_WW_OT", "2_Wen_WS_OT", "2_Wen_WW_HS", "2_Wen_WS_HS","2_Sto_WW_OT", "2_Sto_WS_OT", "2_Sto_WW_HS", "2_Sto_WS_HS"), showGeno = TRUE)p_u3=plot(U3_all_time_point, plotType = "layout", timePoints = c(1), highlight = c("1_Wen_WW_OT", "1_Sto_WW_OT","2_Wen_WW_OT", "2_Wen_WS_OT", "2_Wen_WW_HS", "2_Wen_WS_HS","2_Sto_WW_OT", "2_Sto_WS_OT", "2_Sto_WW_HS", "2_Sto_WS_HS"), showGeno = TRUE)svg(file=here::here("report/intro/plot/layout_unit_U2.svg"),width = 9, height = 9)p_u2dev.off()svg(file=here::here("report/intro/plot/layout_unit_U3.svg"),width = 9, height = 9)p_u3dev.off()# # Check the heatmap of the raw data at one time point# plot(U2_time_point,# plotType = "layout",# timePoints = 1,# traits = "sum_biomass")```::: {#fig-greenhouse layout-ncol="2"}{#fig-U2}{#fig-U3}Layout of Rhizotube and plants in the two greenhouses:::```{r, eval=F}Trait <-"sum_biomass"spatial_correction<-function(Trait,data_x){ pheno <- data_x[[1]] %>% drop_na(Trait) #%>% filter(genotype %in% c( "2_Wen_WS_OT","2_Sto_WS_OT")) %>% mutate(genotype==substr(genotype, 3, 5)) # select the nb of segments: here all the rows and columns nseg = c(nlevels(pheno$colId), nlevels(pheno$rowId)) cat("Time Point: ", timePoint, "\n", file = output_file) # add timePoint number print(timePoint) fit.SpATS <- SpATS(response = Trait, # fit the SpATS model fixed = ~ repId, random = ~ colId + rowId, spatial = ~ PSANOVA(colNum, rowNum, nseg = nseg, nest.div = c(2, 2)), genotype = "genotype", genotype.as.random = TRUE, data = pheno, control = list(maxit = 50, tolerance = 1e-03, monitoring = 0)) summary_output <- capture.output(summary(fit.SpATS)) # Captures model summary output all_summaries <- c(all_summaries, paste0("Time Point: ", timePoint, "\n", summary_output))# Add output to variable writeLines(all_summaries, con = output_file) cat("Traitement pour le trait", Trait, "terminé.\n") plot(fit.SpATS, spaTrend = "percentage")## plot of the spats model with spatial trend as percentage modPhenoSpCheck <- fitModels(TP = data_x, trait = Trait, useRepId = TRUE, useCheck = FALSE, timePoints = 1) px<-plot(modPhenoSpCheck, timePoints = 1, plotType = "spatial", spaTrend = "percentage") ## Extract the corrected values for one time point spatCorrSp <- getCorrected(modPhenoSpCheck, timePoints = timePoint) varianceSp <- getVar(modPhenoSpCheck) ## Extract model components: heritSp <- getHerit(modPhenoSpCheck) effDimSp <- getEffDims(modPhenoSpCheck) return(list(list(plot = px, datacorrect = spatCorrSp)))}result_U2=spatial_correction(Trait = "sum_biomass",data_x = U2_H2_time_point)result_U3=spatial_correction(Trait = "sum_biomass",data_x = U3_H2_time_point)p1<-result_U2[[1]]$plot$`2021-10-11`$p4p2<-result_U3[[1]]$plot$`2021-10-11`$p4p1=p1+labs(title = paste0("Filtted Spatial Trend Unit 2", " ", "sum_biomass"))p2=p2+labs(title = paste0("Filtted Spatial Trend Unit 3" , " ", "sum_biomass"))ggsave(here::here("report/intro/plot/ggcoef_model.svg"),p1+p2)result_all=rbind(result_U2[[1]]$"datacorrect",result_U3[[1]]$"datacorrect")p1=result_all %>% ggplot(aes(x=genotype,y=sum_biomass, fill=genotype))+geom_boxplot()+labs(title="Without correction for sum biomass")p2=result_all %>% ggplot(aes(x=genotype,y=sum_biomass_corr, fill=genotype))+geom_boxplot()+labs(title="With correction for sum biomass")ggsave(here::here("report/intro/plot/comparison.svg"),p1+p2)```</details>






