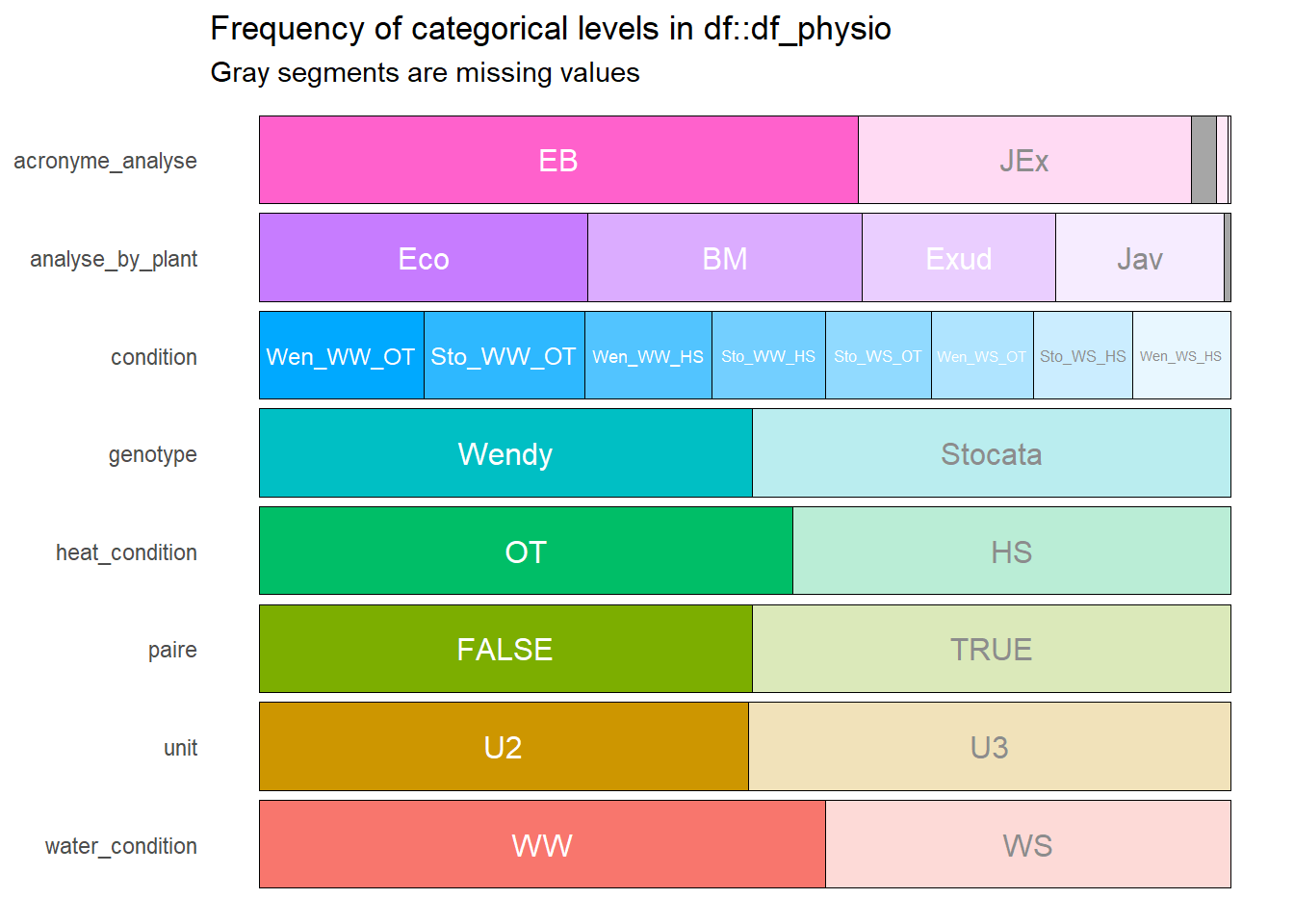
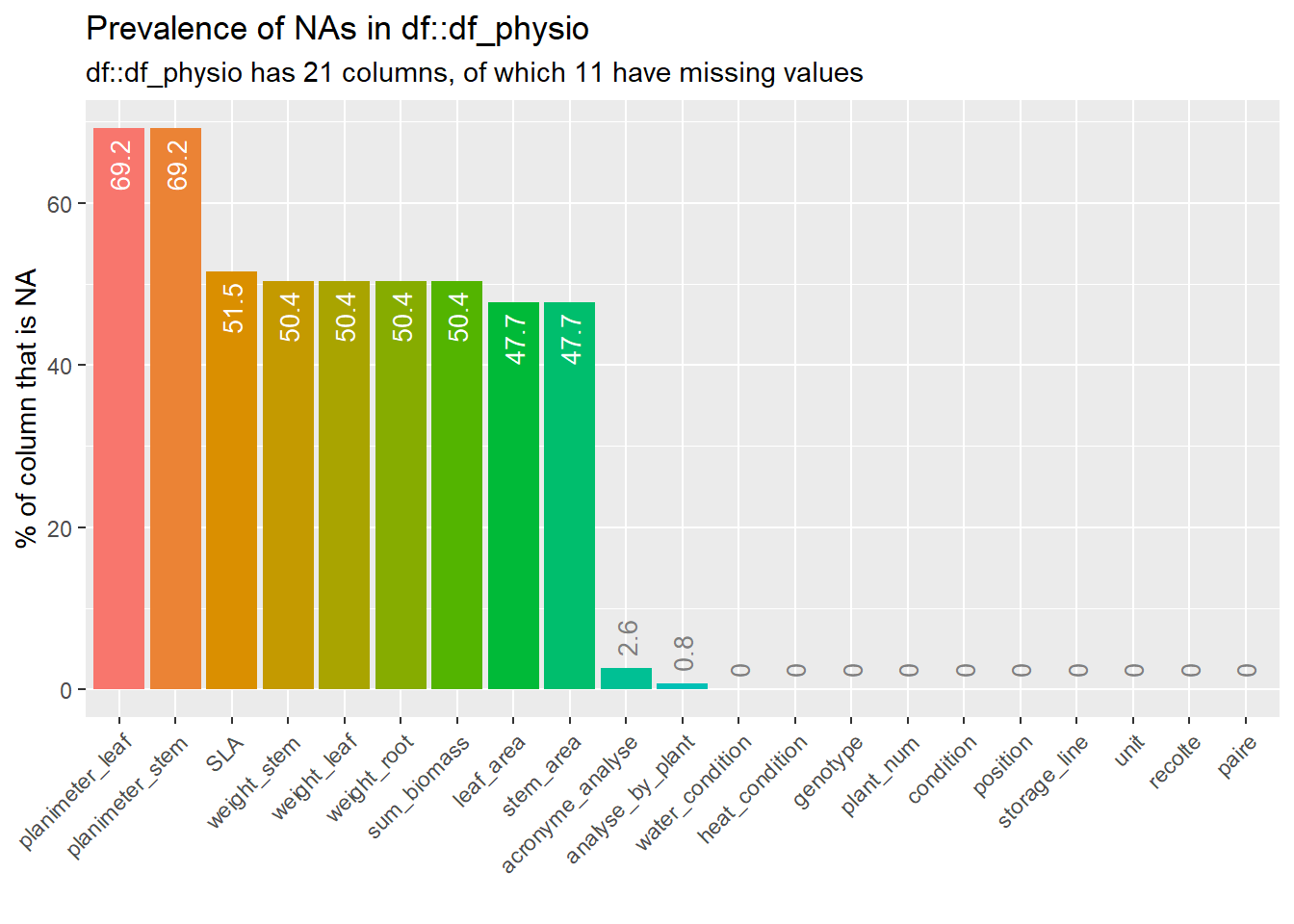
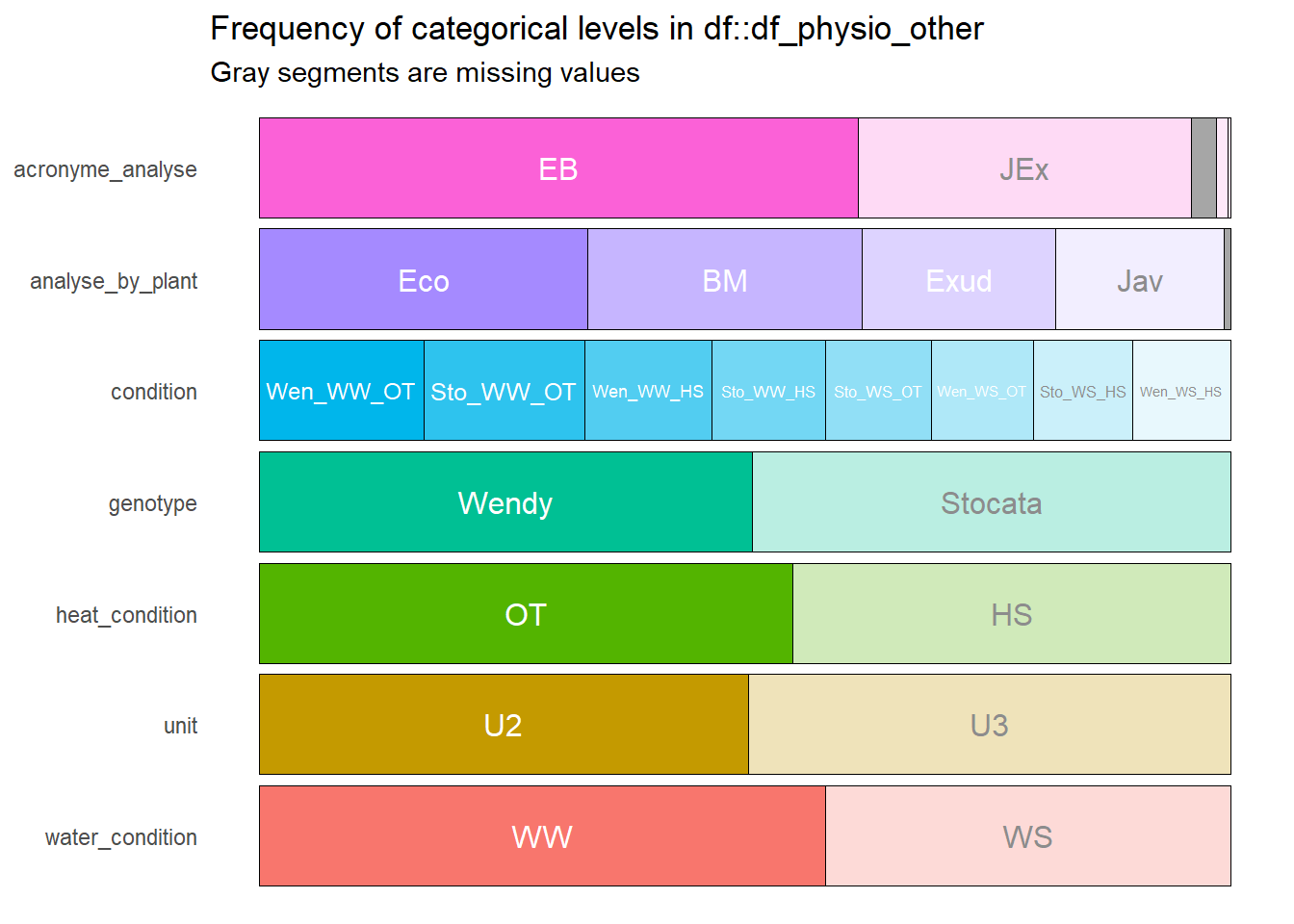
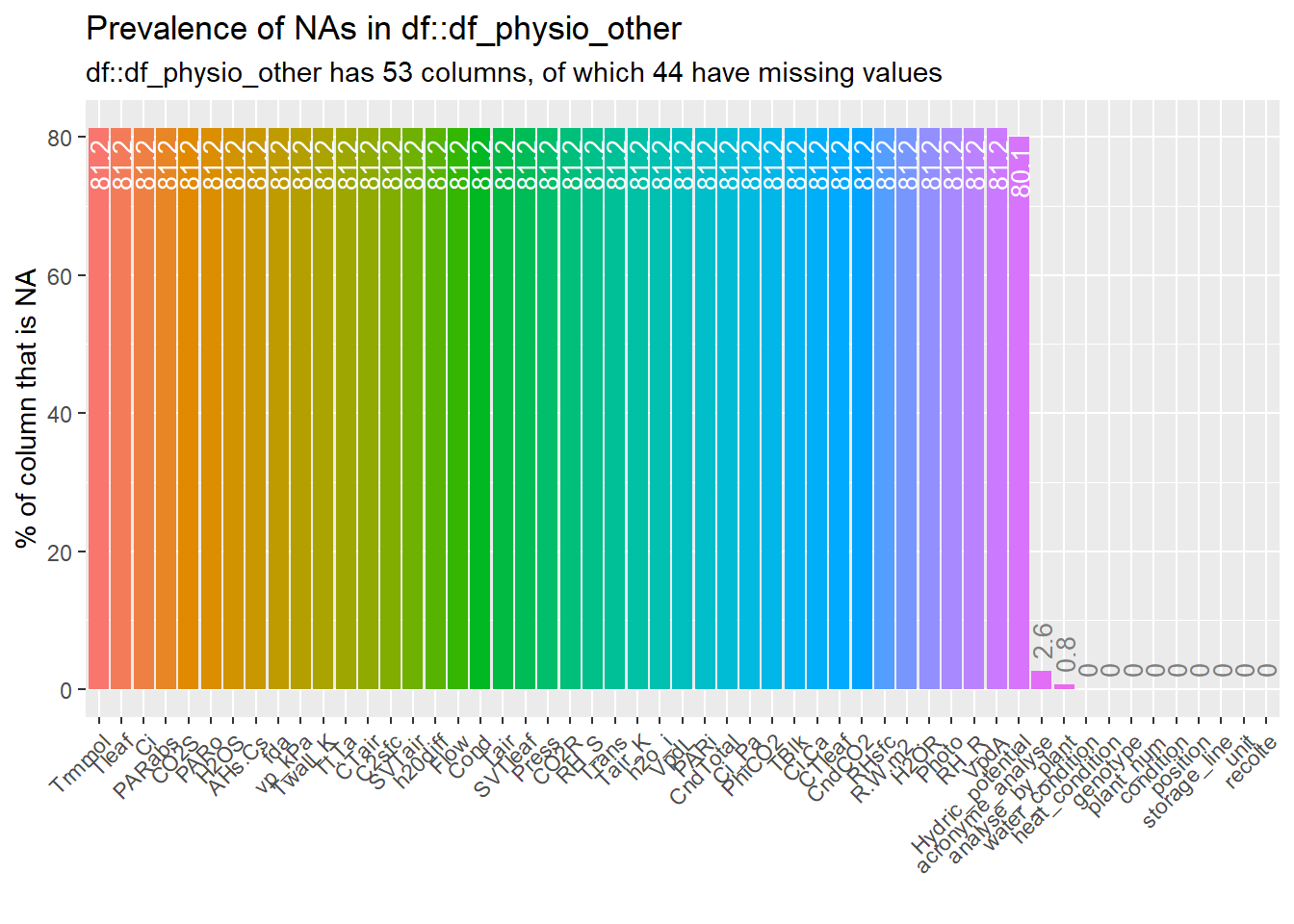
--- output: html_document editor_options: chunk_output_type: console --- ## Rapid analysis of ecophysiological data {#sec-physio_resum} ```{r setup,message=FALSE, warning=FALSE,echo=T} # Pkg ----- #package library(ggplot2) library(plotly) library(readxl) library(tidyverse) library(car) library(Rmisc) library(multcomp) library(lubridate) library(dplyr) library(scales) library(hms) library(tictoc) library(multcompView) library(gridExtra) library(kableExtra) library(cowplot) library(kableExtra) library(ggpubr) library(labelled) library(gtsummary) library(ggstats) library(performance) library(inspectdf) library(patchwork) library(gridExtra) library(ggpubr) library(foreach) library(parallel) library(MASS) library(rstatix) #function source(here::here("src/function/stat_function/stat_analysis_main.R")) # for make plot source(here::here("src/function/graph_function.R")) set.seed(1) # cosmetics climate_pallet=read_excel(here::here("data/color_palette.xlsm")) %>% filter(set == "climat_condition") %>% dplyr::select(color, treatment) %>% pull(color) %>% setNames(read_excel(here::here("data/color_palette.xlsm")) %>% filter(set == "climat_condition") %>% pull(treatment) ) ``` ## For data related to biomass ### Data importation ```{r} <- read.csv2 (here:: here ("data/physio/global_physio.csv" )) %>% mutate (across (c ("genotype" ,"water_condition" ,"heat_condition" ), as.factor)) %>% mutate (across (c ("storage_line" ,"position" ), as.integer)) %>% mutate (water_condition= relevel (water_condition, "WW" )) %>% mutate (heat_condition= relevel (heat_condition, "OT" )) %>% :: select (acronyme_analyse, water_condition, heat_condition, genotype, "plant_num" ,"condition" ,"position" ,"storage_line" ,"unit" ,"analyse_by_plant" , "recolte" , "paire" ,"weight_stem" ,"weight_leaf" ,"weight_root" ,"leaf_area" , "stem_area" , "planimeter_leaf" , "planimeter_stem" , "SLA" , "sum_biomass" ) %>% # dplyr::select(acronyme_analyse, water_condition, heat_condition, genotype, "plant_num" ,"condition","position" ,"storage_line" ,"unit" ,"analyse_by_plant", "recolte", "paire","Hydric_potential","weight_stem","weight_leaf","weight_root" ,"leaf_area", "stem_area", "planimeter_leaf", "planimeter_stem", "SLA", "sum_biomass", "Trmmol","Tleaf", "Ci","PARabs","CO2S", "PARo", # "H2OS", "AHs.Cs","fda", "vp_kPa", "Twall_K","Tl.Ta", "CTair", "C2sfc", "SVTair","h20diff","Flow", "Cond", "Tair", "SVTleaf","Press", "CO2R", "RH_S", "Trans", "Tair_K","h2o_i", "VpdL", "PARi", "CndTotal","Ci_Pa", "PhiCO2","TBlk", "Ci.Ca", "CTleaf","CndCO2","RHsfc", "R.W.m2.","H2OR", "Photo", "RH_R", "VpdA") %>% drop_na (condition,recolte) %>% mutate (condition= factor (condition,levels= c ("Sto_WW_OT" ,"Sto_WS_OT" ,"Sto_WW_HS" ,"Sto_WS_HS" ,"Wen_WW_OT" ,"Wen_WS_OT" ,"Wen_WW_HS" ,"Wen_WS_HS" )))``` ### Fast data visualization ```{r} <- inspect_cat (df_physio)%>% show_plot ()#inspect_cor() returns a tibble containing Pearson’s correlation coefficient, confidence intervals and p-values for pairs of numeric columns. The function combines the functionality of cor() and cor.test() in a more convenient wrapper. #To plot the comparison of the top 20 correlation coefficients for each condition (not show): # px<-inspect_cor(df_physio %>% # # filter(condition==i) %>% # filter(recolte==2)# %>% # #dplyr::select(starts_with("weight")) # ) %>% # slice(1:40) %>% show_plot() # print(px+labs(title = i)) ``` ```{r} inspect_na (df_physio) %>% show_plot ()``` ### Check for missing values ```{r} <- lapply (df_physio, function (x) {<- df_physio$ plant_num[is.na (x)]return (plantid_with_na)``` ```{r, eval=T} df_physio_h2=df_physio %>% filter(recolte==2) %>% filter(plant_num!=1113) %>% filter(!plant_num %in% c(87)) %>% #filter outlier mutate(condition = case_when( condition == "Sto_WW_OT" ~ "Stocata_WW_OT", condition == "Sto_WS_OT" ~ "Stocata_WS_OT", condition == "Sto_WW_HS" ~ "Stocata_WW_HS", condition == "Sto_WS_HS" ~ "Stocata_WS_HS", condition == "Wen_WW_OT" ~ "Wendy_WW_OT", condition == "Wen_WS_OT" ~ "Wendy_WS_OT", condition == "Wen_WW_HS" ~ "Wendy_WW_HS", condition == "Wen_WS_HS" ~ "Wendy_WS_HS", TRUE ~ condition # Keep other values unchanged )) %>% mutate(climat_condition=paste0(water_condition,"_",heat_condition)) %>% mutate(climat_condition=factor(climat_condition,levels=c("WW_OT","WS_OT","WW_HS","WS_HS"))) %>% mutate(weight_AP=weight_stem+weight_leaf) %>% mutate(ratio_s_h=weight_AP/weight_root) ``` ### Find Outlier ```{r, eval=F} var_to_test=c("Hydric_potential","weight_stem","weight_leaf","weight_root" ,"leaf_area", "stem_area", "planimeter_leaf", "planimeter_stem", "SLA", "sum_biomass", "Trmmol","Tleaf", "Ci","PARabs","CO2S", "PARo", "H2OS", "AHs.Cs","fda", "vp_kPa", "Twall_K","Tl.Ta", "CTair", "C2sfc", "SVTair","h20diff","Flow", "Cond", "Tair", "SVTleaf","Press", "CO2R", "RH_S", "Trans", "Tair_K","h2o_i", "VpdL", "PARi", "CndTotal","Ci_Pa", "PhiCO2","TBlk", "Ci.Ca", "CTleaf","CndCO2","RHsfc", "R.W.m2.","H2OR", "Photo", "RH_R", "VpdA") var_to_test=c("weight_stem","weight_leaf","weight_root" ,"leaf_area", "stem_area", "planimeter_leaf", "planimeter_stem", "SLA", "sum_biomass") num_cores <- detectCores() # Use all the cores available on your machine cl=parallel::makeCluster(as.integer(num_cores-1)) doParallel::registerDoParallel(cl) results <- foreach(i = var_to_test, .packages = c("dplyr", "here"), .combine = rbind) %dopar% { source(here::here("src/function/stat_function/stat_analysis_main.R")) # for make plot anova_factor=2 #cat_col(i, "green") plot_x=stat_analyse( data=df_physio_h2 %>% data.frame() %>% drop_na(all_of(i)) %>% drop_na(plant_num, condition) %>% #dplyr::select(genotype, condition, water_condition, heat_condition,plant_num,climat_condition, all_off(i)) %>% mutate(condition = case_when( condition == "Stocata_WW_OT" ~ "Sto_WW_OT", condition == "Stocata_WS_OT" ~ "Sto_WS_OT", condition == "Stocata_WW_HS" ~ "Sto_WW_HS", condition == "Stocata_WS_HS" ~ "Sto_WS_HS", condition == "Wendy_WW_OT" ~ "Wen_WW_OT", condition == "Wendy_WS_OT" ~ "Wen_WS_OT", condition == "Wendy_WW_HS" ~ "Wen_WW_HS", condition == "Wendy_WS_HS" ~ "Wen_WS_HS", TRUE ~ condition # Keep other values unchanged )) %>% mutate(condition=factor(condition,levels=c("Sto_WW_OT","Stoc_WS_OT","Sto_WW_HS","Sto_WS_HS","Wen_WW_OT","Wen_WS_OT","Wen_WW_HS","Wen_WS_HS"))) %>% mutate(climat_condition=factor(climat_condition,levels=c("WW_OT","WS_OT","WW_HS","WS_HS"))), column_value = i, category_variables = c("climat_condition"), grp_var = "genotype", show_plot = F, outlier_show = F, label_outlier = "plant_num", biologist_stats = T, #Ylab_i = paste0("Shoot / Root ratio"), control_conditions = c("WW_OT") ) plot_x[["outliers"]] } parallel::stopCluster(cl) colnames(results) results %>% filter(is.extreme==T) %>% mutate(plant_num=as.factor(plant_num)) %>% count("plant_num") %>% arrange(desc(freq)) results %>% filter(is.outlier==T) %>% mutate(plant_num=as.factor(plant_num)) %>% count("plant_num") %>% arrange(desc(freq)) ``` ## I deleted the plant numbers 1113 and 87 because they are extreme outliers for several variables. ### Biomass representation ```{r,eval=F} df_physio_h2_mean=df_physio_h2%>% drop_na(condition,genotype,climat_condition,weight_leaf,weight_stem, weight_root) %>% dplyr::select(condition,genotype,climat_condition,plant_num,weight_leaf,weight_stem, weight_root) %>% pivot_longer(c(weight_leaf,weight_stem,weight_root), names_to="organe", values_to="biomass") %>% dplyr::group_by(condition, genotype,climat_condition, organe) %>% dplyr::summarise(Mean=mean(biomass),Sd=sd(biomass))%>% mutate(organe = case_when( organe == "weight_leaf" ~ "Leaf", organe == "weight_stem" ~ "Stem", organe == "weight_root" ~ "Root", TRUE ~ organe # Keep other values unchanged )) %>% pivot_wider(names_from = organe, values_from = c("Mean","Sd")) %>% mutate(adj_Leaf=Mean_Leaf + Mean_Stem + Mean_Root) %>% mutate(adj_Stem= Mean_Stem + Mean_Root) %>% mutate(adj_Root= Mean_Root) %>% pivot_longer(cols = !c(condition,genotype,climat_condition), names_to = c('Type','Organe'), names_sep = '_', values_to = 'value') %>% pivot_wider(names_from = Type, values_from = value) %>% dplyr::rename("Dry Biomass"=Mean) %>% mutate(Organe=factor(Organe,levels=c("Leaf","Stem","Root"))) Fig1=ggplot(df_physio_h2_mean, aes(x = Organe,y=`Dry Biomass`*1000,fill=climat_condition))+geom_errorbar(aes(ymin=`Dry Biomass`*1000-Sd*1000, ymax=`Dry Biomass`*1000+Sd*1000), width=.4,position=position_dodge(.9))+geom_bar(stat="identity",position=position_dodge(),color="black")+ theme_minimal()+ geom_text(aes(label = round(`Dry Biomass`*1000, digits = 0),y=2), color = "white",position = position_dodge(0.9),size=4,angle = 90,hjust = 'left')+ scale_x_discrete(limits=c("Leaf", "Stem", "Root"))+ylab("Dry biomass of each organ (mg)")+ xlab("Organe")+facet_grid(.~genotype)+scale_fill_manual(values=manu_palett) print(Fig1) # Add below the sd the letter df_physio_h2_mean$letter=c( "c","e","d", "c","cd","cd", "b","b","b", "ab","a","ab", "c","cd","de", "c","c","c", "a","ab","ab", "ab","a","a" ) mean_letter1=df_physio_h2_mean %>% # Specify data frame group_by(condition,climat_condition,genotype) %>% # Specify group indicator summarise_at(vars(`Dry Biomass`), # Specify column list(sum)) mean_letter2=df_physio_h2_mean %>% # Specify data frame group_by(condition,climat_condition,genotype) %>% filter(Organe=="Leaf")%>%select(Sd) mean_letter1=cbind(mean_letter1,mean_letter2[,4]) mean_letter1$adj_tot=mean_letter1$`Dry Biomass`+mean_letter1$Sd #for letter Plettre_modif(Z=data_biomass_select,Y=data_biomass_select$Node_weight+data_biomass_select$Root_weight+data_biomass_select$PA,X=data_biomass_select$Genotype,Xlab="x",Ylab = "y") mean_letter1$letter=c("d","cd","b","ab","cd","c","ab","a") color_organe <- c("#68a500","#d9d2b1","#ce7f50") #leaf,#stem #root #graph biomass Fig_biomass=ggplot(df_physio_h2_mean, aes(x = climat_condition,y=`Dry Biomass`*1000,fill=Organe))+ geom_text(position = position_stack(vjust = 1.00),data = mean_letter1, aes(y = adj_tot*1010+Sd*1000, label = letter, fill = "Leaf"),fontface = "bold",color="#226896")+ geom_bar(stat = "identity",color="black")+ theme_bw()+ geom_errorbar(aes(ymin=adj*1000, ymax=adj*1000+Sd*1000),width=.4,position="identity")+ geom_text(position = position_stack(vjust = 0.5), aes(label = letter), color = "black",size=4)+ scale_y_continuous(expand = c(0, 0), limits = c(0, 1700)) + # Ensures y-axis starts at zero ylab("Dry biomass of each organ (mg)")+ xlab("Condition")+ scale_fill_manual(values=color_organe)+ theme(axis.text.x = element_text(angle=90, hjust=1, vjust=1))+ labs(fill = "Organ")+ facet_grid(.~genotype)+ theme(#axis.text.x = element_text(angle=45, hjust=1, vjust=1), legend.position = "right", panel.grid = element_blank(), panel.background = element_rect(fill = "white", colour = "black"), panel.border = element_rect(fill = NA, colour = "black"), strip.background = element_rect(color = "black"), axis.title.x=element_blank(), axis.text.x=element_blank(), axis.ticks.x=element_blank() ) plot_x=stat_analyse( data=df_physio_h2 %>% data.frame() %>% drop_na(ratio_s_h,plant_num, condition) %>% dplyr::select(genotype, condition, water_condition, heat_condition, ratio_s_h,plant_num,climat_condition) %>% mutate(condition = case_when( condition == "Stocata_WW_OT" ~ "Sto_WW_OT", condition == "Stocata_WS_OT" ~ "Sto_WS_OT", condition == "Stocata_WW_HS" ~ "Sto_WW_HS", condition == "Stocata_WS_HS" ~ "Sto_WS_HS", condition == "Wendy_WW_OT" ~ "Wen_WW_OT", condition == "Wendy_WS_OT" ~ "Wen_WS_OT", condition == "Wendy_WW_HS" ~ "Wen_WW_HS", condition == "Wendy_WS_HS" ~ "Wen_WS_HS", TRUE ~ condition # Keep other values unchanged )) %>% mutate(condition=factor(condition,levels=c("Sto_WW_OT","Stoc_WS_OT","Sto_WW_HS","Sto_WS_HS","Wen_WW_OT","Wen_WS_OT","Wen_WW_HS","Wen_WS_HS"))) %>% mutate(climat_condition=factor(climat_condition,levels=c("WW_OT","WS_OT","WW_HS","WS_HS"))), column_value = "ratio_s_h", category_variables = c("climat_condition"), grp_var = "genotype", show_plot = T, outlier_show = F, label_outlier = "plant_num", biologist_stats = T, Ylab_i = paste0("Shoot / Root ratio"), control_conditions = c("WW_OT"), hex_pallet = climate_pallet ) plot_c=Fig_biomass+plot_x[["plot"]]+plot_layout(ncol = 1,heights = c(2.8, 1))+ plot_annotation(tag_levels = 'A') plot_c=ggarrange(Fig_biomass,plot_x[["plot"]], widths = c(1, 1),heights = c(1.7, 1),ncol=1, nrow=2,labels = c("A","B"),align='v') ggsave(here::here("report/physio/plot/biomass_ratio_h2.svg"), plot_c, height = 7.5,width = 6) ```  ### Multiple regression model ```{r,eval=T} df_physio_h2<-read.csv2(here::here("data/physio/global_physio.csv")) %>% mutate(across(c("genotype","water_condition","heat_condition"), as.factor)) %>% mutate(across(c("storage_line","position"), as.integer)) %>% mutate(water_condition=relevel(water_condition, "WW")) %>% mutate(heat_condition=relevel(heat_condition, "OT")) %>% filter(recolte==2) %>% mutate(shoot_root=(weight_stem+weight_leaf)/weight_root) contrasts(df_physio_h2$genotype) <- contr.sum # to say look at the big average only for genotype (juste an other representation) mod1=lm(formula = shoot_root ~ water_condition*heat_condition*genotype+position+storage_line, data = df_physio_h2 %>% drop_na(shoot_root, position),contrasts = list(genotype = MASS::contr.sdif)) p_x<-ggcoef_model(mod1) ggsave(here::here("report/physio/plot/ggcoef_model_1.svg"),p_x) ``` ```{r, warning=FALSE, message=FALSE,echo=F,include = FALSE} mod2 <- step(mod1) p_x<-ggcoef_model(mod2) ggsave(here::here("report/physio/plot/ggcoef_model_2.svg"),p_x) ```   ```{r, eval=F} # mod2 |> performance::model_performance() performance::compare_performance(mod1, mod2) mod2 |> tbl_regression() |> bold_labels() |> add_glance_source_note() ``` ```{r, eval=F} ggstats::ggcoef_compare( list(mod1,mod2), tidy_fun = broom.helpers::tidy_marginal_predictions, type = "dodge", vline = FALSE ) # Draft df_physio_h2 %>% dplyr::select(plant_num,water_condition, heat_condition, genotype, sum_biomass) %>% group_by(water_condition, heat_condition, genotype) %>% identify_outliers(sum_biomass) df_without_outlier<-df_physio_h2 %>% filter(!plant_num%in% c(143,1063,1091,1129)) %>% drop_na(sum_biomass) m_out<-lm(data=df_without_outlier,sum_biomass~genotype*heat_condition*water_condition) ggqqplot(residuals(m_out)) shapiro_test(residuals(m_out)) ggqqplot(df_physio_h2, "sum_biomass", ggtheme = theme_bw()) + facet_grid(water_condition + heat_condition ~ genotype, labeller = "label_both") df_physio_h2 %>% levene_test(sum_biomass ~ water_condition*heat_condition*genotype) summary(mod2) res.aov <- df_physio_h2 %>% anova_test(sum_biomass ~ water_condition*heat_condition*genotype) res.aov res.aov2 <- df_physio_h2 %>% welch_anova_test(sum_biomass ~ condition) # Pairwise comparisons (Games-Howell) pwc2 <- df_physio_h2 %>% games_howell_test(sum_biomass ~ condition) # Visualization: Boxplots with p-values mod=rlm(data=df_without_outlier,sum_biomass~heat_condition*water_condition) summary(mod) anova(mod) hist(df_without_outlier$sum_biomass, main = "Histogram of my data", xlab = "Valeurs", ylab = "Frequency", col = "skyblue", border = "white") test=df_physio_h2 %>% tukey_hsd(sum_biomass ~ genotype*water_condition*heat_condition) ``` ### Contribution of each variable related to biomass ```{r, eval=F} df_physio_h2_v<-df_physio_h2 %>% dplyr::select(plant_num,genotype,heat_condition,water_condition,sum_biomass,weight_leaf, weight_stem, weight_root,ratio_s_h,"leaf_area") %>% pivot_longer(-c(plant_num,genotype,heat_condition,water_condition),names_to = "variable") compile_result=as.data.frame(matrix(data=NA,nrow = 0, ncol = 8)) for (i in 1:length(levels(as.factor(df_physio_h2_v$variable)))){ variable_l_i=as.character(levels(as.factor(df_physio_h2_v$variable))[i]) cat( "Variable:" ,variable_l_i, "\n") df_x= df_physio_h2_v %>% filter(variable==variable_l_i) %>% drop_na(value) formula_string <- as.formula(paste("value", "~", paste("genotype","*","water_condition","*","heat_condition", sep = ""))) # Perform ANOVA using the aov function result_variable <- aov(data = df_x, formula = formula_string) anova_result=summary(result_variable)[[1]] %>% mutate(R2 = `Sum Sq` / sum(`Sum Sq`)) row_name<-rownames(anova_result) row_name <- gsub("genotype","Genotype",row_name) row_name <- gsub("water_condition","Water",row_name) row_name <- gsub("heat_condition","Heat",row_name) rownames(anova_result)<-row_name anova_result$variable=as.character(df_x$variable)[1] compile_result=rbind(compile_result,anova_result) } compile_result<-compile_result %>% as.data.frame() %>% rownames_to_column("contribution_variable") %>% mutate(Significance = case_when( `Pr(>F)` <= 0.001 ~ '***', `Pr(>F)` < 0.01 ~ '**', `Pr(>F)` < 0.05 ~ '*', `Pr(>F)` < 0.1 ~ '.', TRUE ~ ' ' )) compile_result=compile_result%>% filter(str_detect(contribution_variable, "Total", negate = TRUE)) %>% filter(str_detect(contribution_variable, "Residual", negate = TRUE)) %>% mutate(contribution_variable = str_replace_all(contribution_variable, " ", "")) %>% mutate_at(vars(contribution_variable), ~str_replace_all(., "\\$", ""))%>% mutate_at(vars(contribution_variable), ~str_replace_all(., "[0-9]", "")) %>% mutate(contribution_variable = as.character(contribution_variable)) %>% mutate(fontcolor = ifelse(contribution_variable %in% c("Water", "Genotype:Water:Heat"), "#ffffff", "#000000")) %>% mutate(contribution_variable=fct_relevel(contribution_variable,c("Genotype","Water","Heat", "Genotype:Water","Genotype:Heat","Water:Heat","Genotype:Water:Heat"))) %>% mutate(text_output=paste0(round(R2*100,0), "% ", Significance)) plot_contrib<-ggplot(compile_result, aes(x = variable, y = R2, fill = contribution_variable)) + geom_bar(stat = "identity") + geom_text(aes(label = ifelse(R2 > 0.02, text_output, "")), color = compile_result$fontcolor, position = position_stack(vjust = 0.5), size = 2.5)+ scale_fill_manual(values = c("#ffd166", # geno "#118ab2", # water "#ef476f", # heat "#06d6a0", # watergeno "#f78c6b", # genoheat "#FFB6C1", # waterheat "#333333"), name = "title of legend (contribution") +# all scale_y_continuous(labels = scales::percent_format())+ scale_color_identity()+ labs(x = "Compartment", y = "The relative contribution for the different variables (%)", ) + theme_minimal()+ theme(panel.grid = element_blank(), legend.title = element_blank(), # for delete title axis.text.x = element_text(angle = 45,hjust = 1, vjust = 1), axis.title.x = element_blank() ) ggsave(here::here("report/physio/plot/contribution.svg"),plot_contrib) ```  ### Student test ```{r, eval=F} test=read.csv2(here::here("data/physio/global_physio.csv")) %>% mutate(across(c("genotype","water_condition","heat_condition"), as.factor)) %>% mutate(across(c("storage_line","position"), as.integer)) %>% mutate(water_condition=relevel(water_condition, "WW")) %>% mutate(heat_condition=relevel(heat_condition, "OT")) %>% filter(recolte==2) %>% drop_na(Hydric_potential) %>% #filter(climat_condition=="WW_OT") %>% t_test(Hydric_potential ~ condition,p.adjust.method = "BH") %>% add_significance() ``` ```{r,eval=F} df_physio_s=read.csv2(here::here("data/physio/global_physio.csv")) %>% mutate(across(c("genotype","water_condition","heat_condition"), as.factor)) %>% mutate(across(c("storage_line","position"), as.integer)) %>% mutate(water_condition=relevel(water_condition, "WW")) %>% mutate(heat_condition=relevel(heat_condition, "OT")) %>% filter(recolte==2) %>% drop_na(Hydric_potential) %>% #filter(genotype=="Stocata") %>% #filter(climat_condition%in% c("WW_OT","WS_HS")) %>% dplyr::group_by(condition) %>% dplyr::summarise(Mean=mean(Hydric_potential)) (df_physio_s$Mean[1]-df_physio_s$Mean[2])/df_physio_s$Mean[2] ``` ## For the data related to licor or leaf water pottential ### Data importation ```{r} <- read.csv2 (here:: here ("data/physio/global_physio.csv" )) %>% mutate (across (c ("genotype" ,"water_condition" ,"heat_condition" ), as.factor)) %>% mutate (across (c ("storage_line" ,"position" ), as.integer)) %>% mutate (water_condition= relevel (water_condition, "WW" )) %>% mutate (heat_condition= relevel (heat_condition, "OT" )) %>% :: select (acronyme_analyse, water_condition, heat_condition, genotype, "plant_num" ,"condition" ,"position" ,"storage_line" ,"unit" ,"analyse_by_plant" , "recolte" , #"paire","weight_stem","weight_leaf","weight_root" ,"leaf_area", "stem_area", "planimeter_leaf", "planimeter_stem", "SLA", "sum_biomass") %>% # dplyr::select(acronyme_analyse, water_condition, heat_condition, genotype, "plant_num" ,"condition","position" ,"storage_line" ,"unit" ,"analyse_by_plant", "recolte", "Hydric_potential" , "Trmmol" ,"Tleaf" , "Ci" ,"PARabs" ,"CO2S" , "PARo" , "H2OS" , "AHs.Cs" ,"fda" , "vp_kPa" , "Twall_K" ,"Tl.Ta" , "CTair" , "C2sfc" , "SVTair" ,"h20diff" ,"Flow" , "Cond" , "Tair" , "SVTleaf" ,"Press" , "CO2R" , "RH_S" , "Trans" , "Tair_K" ,"h2o_i" , "VpdL" , "PARi" , "CndTotal" ,"Ci_Pa" , "PhiCO2" ,"TBlk" , "Ci.Ca" , "CTleaf" ,"CndCO2" ,"RHsfc" , "R.W.m2." ,"H2OR" , "Photo" , "RH_R" , "VpdA" ) %>% drop_na (condition,recolte) %>% mutate (condition= factor (condition,levels= c ("Sto_WW_OT" ,"Sto_WS_OT" ,"Sto_WW_HS" ,"Sto_WS_HS" ,"Wen_WW_OT" ,"Wen_WS_OT" ,"Wen_WW_HS" ,"Wen_WS_HS" )))``` ### Fast data visualization ```{r} <- inspect_cat (df_physio_other)%>% show_plot ()``` ```{r, eval=F} #inspect_cor() returns a tibble containing Pearson’s correlation coefficient, confidence intervals and p-values for pairs of numeric columns. The function combines the functionality of cor() and cor.test() in a more convenient wrapper. #To plot the comparison of the top 20 correlation coefficients for each condition (not show): # px<-inspect_cor(df_physio %>% # # filter(condition==i) %>% # filter(recolte==2)# %>% # #dplyr::select(starts_with("weight")) # ) %>% # slice(1:40) %>% show_plot() # print(px+labs(title = i)) ``` ```{r} inspect_na (df_physio_other) %>% show_plot ()``` ### Check for missing values ```{r} <- lapply (df_physio_other, function (x) {<- df_physio_other$ plant_num[is.na (x)]return (plantid_with_na)``` ```{r, eval=T} df_physio_other_h2=df_physio_other %>% filter(recolte==2) %>% filter(plant_num!=1113) %>% filter(!plant_num %in% c(87)) %>% #filter outlier mutate(condition = case_when( condition == "Sto_WW_OT" ~ "Stocata_WW_OT", condition == "Sto_WS_OT" ~ "Stocata_WS_OT", condition == "Sto_WW_HS" ~ "Stocata_WW_HS", condition == "Sto_WS_HS" ~ "Stocata_WS_HS", condition == "Wen_WW_OT" ~ "Wendy_WW_OT", condition == "Wen_WS_OT" ~ "Wendy_WS_OT", condition == "Wen_WW_HS" ~ "Wendy_WW_HS", condition == "Wen_WS_HS" ~ "Wendy_WS_HS", TRUE ~ condition # Keep other values unchanged )) %>% mutate(climat_condition=paste0(water_condition,"_",heat_condition)) %>% mutate(climat_condition=factor(climat_condition,levels=c("WW_OT","WS_OT","WW_HS","WS_HS"))) ``` ### Contribution of each variable related to other variable ```{r,eval=F} df_physio_h2_v=df_physio_other_h2 %>% dplyr::select(-c(acronyme_analyse,position, storage_line, unit, analyse_by_plant, recolte, Flow,fda,Press, CO2R,RH_R)) %>% # not interesting data measured by lycor pivot_longer(-c(plant_num,genotype,heat_condition,water_condition,condition,climat_condition),names_to = "variable",values_to = "value") compile_result=as.data.frame(matrix(data=NA,nrow = 0, ncol = 8)) for (i in 1:length(levels(as.factor(df_physio_h2_v$variable)))){ variable_l_i=as.character(levels(as.factor(df_physio_h2_v$variable))[i]) cat( "Variable:" ,variable_l_i, "\n") df_x= df_physio_h2_v %>% filter(variable==variable_l_i) %>% drop_na(value) formula_string <- as.formula(paste("value", "~", paste("genotype","*","water_condition","*","heat_condition", sep = ""))) # Carry out ANOVA using the aov function result_variable <- aov(data = df_x, formula = formula_string) anova_result=summary(result_variable)[[1]] %>% mutate(R2 = `Sum Sq` / sum(`Sum Sq`)) row_name<-rownames(anova_result) row_name <- gsub("genotype","Genotype",row_name) row_name <- gsub("water_condition","Water",row_name) row_name <- gsub("heat_condition","Heat",row_name) rownames(anova_result)<-row_name anova_result$variable=as.character(df_x$variable)[1] compile_result=rbind(compile_result,anova_result) } compile_result<-compile_result %>% as.data.frame() %>% rownames_to_column("contribution_variable") %>% mutate(Significance = case_when( `Pr(>F)` <= 0.001 ~ '***', `Pr(>F)` < 0.01 ~ '**', `Pr(>F)` < 0.05 ~ '*', `Pr(>F)` < 0.1 ~ '.', TRUE ~ ' ' )) compile_result=compile_result%>% filter(str_detect(contribution_variable, "Total", negate = TRUE)) %>% filter(str_detect(contribution_variable, "Residual", negate = TRUE)) %>% mutate(contribution_variable = str_replace_all(contribution_variable, " ", "")) %>% mutate_at(vars(contribution_variable), ~str_replace_all(., "\\$", ""))%>% mutate_at(vars(contribution_variable), ~str_replace_all(., "[0-9]", "")) %>% mutate(contribution_variable = as.character(contribution_variable)) %>% mutate(fontcolor = ifelse(contribution_variable %in% c("Water", "Genotype:Water:Heat"), "#ffffff", "#000000")) %>% mutate(contribution_variable=fct_relevel(contribution_variable,c("Genotype","Water","Heat", "Genotype:Water","Genotype:Heat","Water:Heat","Genotype:Water:Heat"))) %>% mutate(text_output=paste0(round(R2*100,0), "% ", Significance)) plot_contrib<-ggplot(compile_result, aes(x = variable, y = R2, fill = contribution_variable)) + geom_bar(stat = "identity") + geom_text(aes(label = ifelse(R2 > 0.02, text_output, "")), color = compile_result$fontcolor, position = position_stack(vjust = 0.5), size = 2.5)+ scale_fill_manual(values = c("#ffd166", # geno "#118ab2", # water "#ef476f", # heat "#06d6a0", # watergeno "#f78c6b", # genoheat "#FFB6C1", # waterheat "#333333"), name = "title of legend (contribution") +# all scale_y_continuous(labels = scales::percent_format())+ scale_color_identity()+ labs(x = "Compartment", y = "The relative contribution for the different variables (%)", ) + theme_minimal()+ theme(panel.grid = element_blank(), legend.title = element_blank(), # for delete title axis.text.x = element_text(angle = 45,hjust = 1, vjust = 1), axis.title.x = element_blank() )+ coord_flip(); plot_contrib ggsave(here::here("report/physio/plot/contribution_other.svg"),plot_contrib, height = 16,width = 10) ```  ### Summary of some interesting variables in ecophysiology #### Plot ```{r, eval=FALSE} df_evapo_H2 <- read.csv(here::here("data/water/weighing_watering_output/sum_evapotranspi.csv"))[-1] %>% dplyr::rename(plant_num = plant_id) %>% mutate(sum_tot_evapo_plant = sum_total_evapotranspiration/2) %>% filter(recolte == 2) %>% dplyr::select(plant_num, sum_tot_evapo_plant) df_physio_other_H2<-read.csv2(here::here("data/physio/global_physio.csv")) %>% left_join(.,df_evapo_H2, by = "plant_num") %>% mutate(across(c("genotype","water_condition","heat_condition"), as.factor)) %>% mutate(across(c("storage_line","position"), as.integer)) %>% mutate(water_condition=relevel(water_condition, "WW")) %>% mutate(heat_condition=relevel(heat_condition, "OT")) %>% mutate(climat_condition=paste0(water_condition,"_",heat_condition)) %>% mutate(climat_condition=factor(climat_condition,levels=c("WW_OT","WS_OT","WW_HS","WS_HS"))) %>% mutate(condition=factor(condition,levels=c("Sto_WW_OT","Sto_WS_OT","Sto_WW_HS","Sto_WS_HS","Wen_WW_OT","Wen_WS_OT","Wen_WW_HS","Wen_WS_HS"))) %>% # dplyr::select(acronyme_analyse, water_condition, heat_condition, genotype, "plant_num" ,"condition","position" ,"storage_line" ,"unit" ,"analyse_by_plant", "recolte", #"paire","weight_stem","weight_leaf","weight_root" ,"leaf_area", "stem_area", "planimeter_leaf", "planimeter_stem", "SLA", "sum_biomass") %>% # # dplyr::select(acronyme_analyse, water_condition, heat_condition, genotype, "plant_num" ,"condition","position" ,"storage_line" ,"unit" ,"analyse_by_plant", "recolte", # "Hydric_potential", "Trmmol","Tleaf", "Ci","PARabs","CO2S", "PARo", "H2OS", "AHs.Cs","fda", "vp_kPa", "Twall_K","Tl.Ta", "CTair", "C2sfc", "SVTair","h20diff","Flow", "Cond", "Tair", "SVTleaf","Press", "CO2R", "RH_S", "Trans", "Tair_K","h2o_i", "VpdL", "PARi", "CndTotal","Ci_Pa", "PhiCO2","TBlk", "Ci.Ca", "CTleaf","CndCO2","RHsfc", "R.W.m2.","H2OR", "Photo", "RH_R", "VpdA") %>% drop_na(condition,recolte) %>% filter(recolte==2) Fig_evapo=stat_analyse( data=df_physio_other_H2 %>% drop_na(climat_condition,sum_tot_evapo_plant), column_value = "sum_tot_evapo_plant", category_variables = c("climat_condition"), grp_var = "genotype", show_plot = T, outlier_show = F, label_outlier = "plant_num", biologist_stats = T, Ylab_i = "Total evapotranspiration (mL)", control_conditions = c("WW_OT"), strip_normale = F, hex_pallet = climate_pallet ) Fig_tleaf=stat_analyse( data=df_physio_other_H2 %>% drop_na(climat_condition,Tleaf), column_value = "Tleaf", category_variables = c("climat_condition"), grp_var = "genotype", show_plot = T, outlier_show = F, label_outlier = "plant_num", biologist_stats = T, Ylab_i = "Leaf temperature (°C)", control_conditions = c("WW_OT"), strip_normale = F, hex_pallet = climate_pallet ) Fig_photo=stat_analyse( data=df_physio_other_H2 %>% drop_na(climat_condition,Photo), column_value = "Photo", category_variables = c("climat_condition"), grp_var = "genotype", show_plot = T, outlier_show = F, label_outlier = "plant_num", biologist_stats = T, Ylab_i = expression("Photosynthesis" ~ mu * mol ~ CO[2] ~ m^-2 ~ s^-1), control_conditions = c("WW_OT"), strip_normale = F, hex_pallet = climate_pallet ) Fig_SLA=stat_analyse( data=df_physio_other_H2 %>% drop_na(climat_condition,SLA), column_value = "SLA", category_variables = c("climat_condition"), grp_var = "genotype", show_plot = T, outlier_show = F, label_outlier = "plant_num", biologist_stats = T, Ylab_i = "Specific leaf area (m²/kg)", control_conditions = c("WW_OT"), strip_normale = F, hex_pallet = climate_pallet ) Fig_LWP=stat_analyse( data=df_physio_other_H2 %>% drop_na(climat_condition,Hydric_potential), column_value = "Hydric_potential", category_variables = c("climat_condition"), grp_var = "genotype", show_plot = T, outlier_show = F, label_outlier = "plant_num", biologist_stats = T, Ylab_i = "Leaf water potential (Mpa)", control_conditions = c("WW_OT"), strip_normale = F, hex_pallet = climate_pallet ) px=ggarrange( Fig_evapo[["plot"]]+labs(color="Treatment",fill="Treatment"), Fig_tleaf[["plot"]]+labs(color="Treatment",fill="Treatment"), Fig_photo[["plot"]]+labs(color="Treatment",fill="Treatment"), #Fig_SLA[["plot"]]+labs(color="Treatment",fill="Treatment"), Fig_LWP[["plot"]]+labs(color="Treatment",fill="Treatment"), ncol=4, nrow=1,common.legend = TRUE, legend="right", #labels = c("A","B","C", "D", "F"), align='h') px #export to delet png(here::here("report/physio/plot/evapo_tleaf_lwp.png"), width = 30, height = 8, units = 'cm', res = 900) grid.arrange (px) # Make plot dev.off() # test=df_physio_other_H2 %>%drop_na(SLA) %>% dplyr::select(plant_num,condition,leaf_scan,planimeter_leaf, leaf_area,weight_leaf,SLA) %>% mutate(scan=ifelse(is.na(planimeter_leaf),"scan","planimeter")) %>% mutate(cond_type=paste0(condition,"_",scan)) %>% # ggplot(aes(x=cond_type,y=leaf_area))+geom_boxplot() ``` 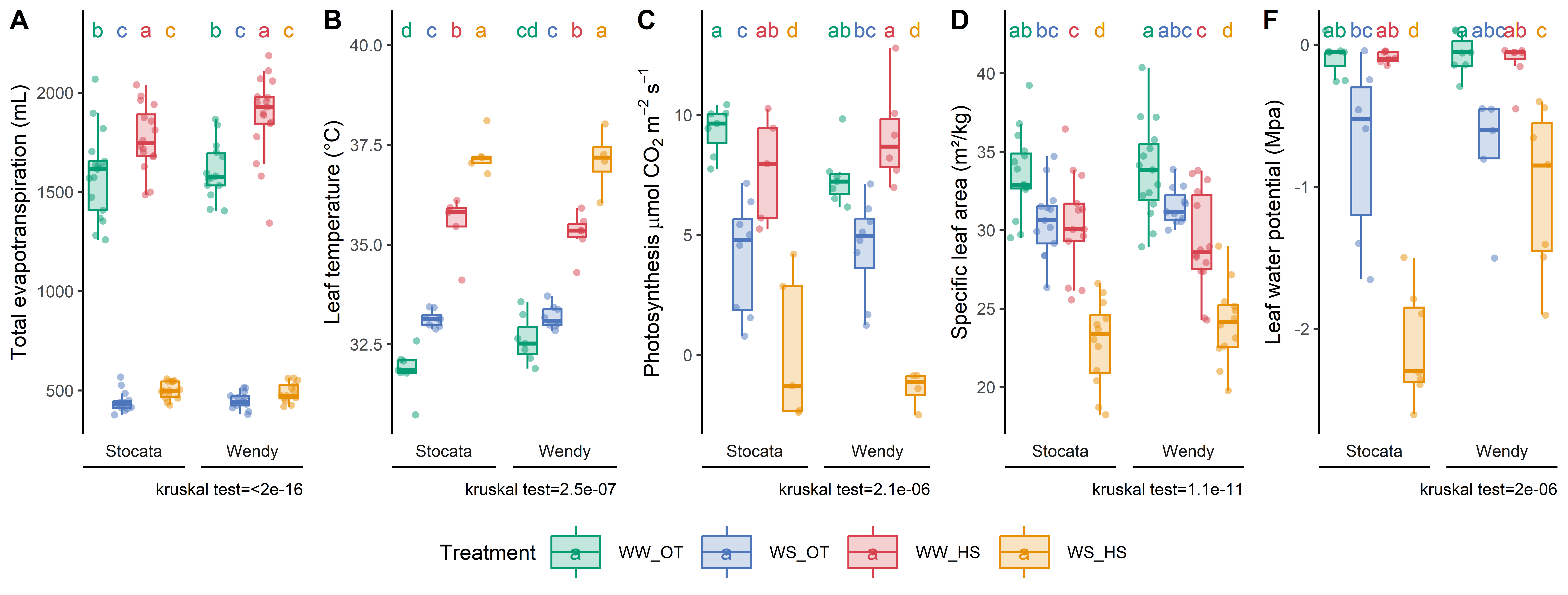 #### Contribution ```{r, eval=F} df_evapo_H2 <- read.csv(here::here("data/water/weighing_watering_output/sum_evapotranspi.csv"))[-1] %>% dplyr::rename(plant_num = plant_id) %>% mutate(sum_tot_evapo_plant = sum_total_evapotranspiration/2) %>% filter(recolte == 2) %>% dplyr::select(plant_num, sum_tot_evapo_plant) df_physio_other_H2_v<-read.csv2(here::here("data/physio/global_physio.csv")) %>% left_join(.,df_evapo_H2, by = "plant_num") %>% mutate(across(c("genotype","water_condition","heat_condition"), as.factor)) %>% mutate(across(c("storage_line","position"), as.integer)) %>% mutate(water_condition=relevel(water_condition, "WW")) %>% mutate(heat_condition=relevel(heat_condition, "OT")) %>% mutate(climat_condition=paste0(water_condition,"_",heat_condition)) %>% mutate(climat_condition=factor(climat_condition,levels=c("WW_OT","WS_OT","WW_HS","WS_HS"))) %>% mutate(condition=factor(condition,levels=c("Sto_WW_OT","Sto_WS_OT","Sto_WW_HS","Sto_WS_HS","Wen_WW_OT","Wen_WS_OT","Wen_WW_HS","Wen_WS_HS"))) %>% drop_na(condition,recolte) %>% filter(recolte==2) %>% dplyr::select(water_condition, heat_condition, genotype, plant_num, condition, climat_condition, sum_tot_evapo_plant, Tleaf,Photo,#SLA, Hydric_potential) %>% pivot_longer(-c(plant_num,genotype,heat_condition,water_condition,condition,climat_condition),names_to = "variable",values_to = "value") %>% mutate(variable = case_when( variable == "sum_tot_evapo_plant" ~ "Total evapotranspiration", variable == "Tleaf" ~ "Leaf temperature", variable == "Photo" ~ "Photosynthesis", variable == "SLA" ~ "Specific leaf area", variable == "Hydric_potential" ~ "Leaf water potential", TRUE ~ variable ) ) compile_result=as.data.frame(matrix(data=NA,nrow = 0, ncol = 8)) for (i in 1:length(levels(as.factor(df_physio_other_H2_v$variable)))){ variable_l_i=as.character(levels(as.factor(df_physio_other_H2_v$variable))[i]) cat( "Variable:" ,variable_l_i, "\n") df_x= df_physio_other_H2_v %>% filter(variable==variable_l_i) %>% drop_na(value) formula_string <- as.formula(paste("value", "~", paste("genotype","*","water_condition","*","heat_condition", sep = ""))) # Carry out ANOVA using the aov function result_variable <- aov(data = df_x, formula = formula_string) anova_result=summary(result_variable)[[1]] %>% mutate(R2 = `Sum Sq` / sum(`Sum Sq`)) row_name<-rownames(anova_result) row_name <- gsub("genotype","Genotype",row_name) row_name <- gsub("water_condition","Water",row_name) row_name <- gsub("heat_condition","Heat",row_name) rownames(anova_result)<-row_name anova_result$variable=as.character(df_x$variable)[1] compile_result=rbind(compile_result,anova_result) } compile_result<-compile_result %>% as.data.frame() %>% rownames_to_column("contribution_variable") %>% mutate(Significance = case_when( `Pr(>F)` <= 0.001 ~ '***', `Pr(>F)` < 0.01 ~ '**', `Pr(>F)` < 0.05 ~ '*', `Pr(>F)` < 0.1 ~ '.', TRUE ~ ' ' )) library(forcats) compile_result=compile_result%>% filter(str_detect(contribution_variable, "Total", negate = TRUE)) %>% filter(str_detect(contribution_variable, "Residual", negate = TRUE)) %>% mutate(contribution_variable = str_replace_all(contribution_variable, " ", "")) %>% mutate_at(vars(contribution_variable), ~str_replace_all(., "\\$", ""))%>% mutate_at(vars(contribution_variable), ~str_replace_all(., "[0-9]", "")) %>% mutate(contribution_variable = as.character(contribution_variable)) %>% mutate(fontcolor = ifelse(contribution_variable %in% c("Water", "Genotype:Water:Heat"), "#ffffff", "#000000")) %>% mutate(contribution_variable=fct_relevel(contribution_variable,c("Genotype","Water","Heat", "Genotype:Water","Genotype:Heat","Water:Heat","Genotype:Water:Heat"))) %>% mutate(text_output=paste0(round(R2*100,0), "% ", Significance)) %>% mutate(variable=factor(variable,levels=c("Total evapotranspiration","Leaf temperature","Photosynthesis","Leaf water potential"))) %>% arrange(desc(variable)) plot_contrib<-ggplot(compile_result, aes(x = fct_inorder(variable), y = R2, fill = contribution_variable)) + geom_bar(stat = "identity") + geom_text(aes(label = ifelse(R2 > 0.02, text_output, "")), color = compile_result$fontcolor, position = position_stack(vjust = 0.5), size = 2.5)+ scale_fill_manual(values = c("#ffd166", # geno "#118ab2", # water "#ef476f", # heat "#06d6a0", # watergeno "#f78c6b", # genoheat "#FFB6C1", # wate rheat "#333333"), name = "title of legend (contribution") +# all scale_y_continuous(labels = scales::percent_format())+ scale_color_identity()+ labs(x = "Variable", y = "The relative contribution for the different variables (%)", ) + theme_classic()+ theme(panel.grid = element_blank(), legend.title = element_blank(), # for delete title axis.text.x = element_text(angle = 0,hjust = 1, vjust = 1), axis.title.x = element_blank(), axis.line = element_line(size = 0.3) )+ coord_flip(); plot_contrib ggsave(here::here("report/physio/plot/contrib_physio_interesting_var.svg"),plot_contrib, height = 2,width = 11) ggsave(here::here("report/physio/plot/contrib_physio_interesting_var.png"),plot_contrib, height = 2,width = 11, dpi = 600) ```